Math Is Fun Forum
You are not logged in.
- Topics: Active | Unanswered
Pages: 1
#1 2025-01-20 16:32:30
- Jai Ganesh
- Administrator

- Registered: 2005-06-28
- Posts: 53,831
Protein Sequencing
Protein Sequencing
Gist
Protein sequencing is the process of determining the amino acid order in a protein chain, revealing its primary structure. Single-molecule protein sequencing is an advanced technique that involves analyzing single amino acid or protein molecules, enabling high-resolution insights into complex biological systems.
Summary
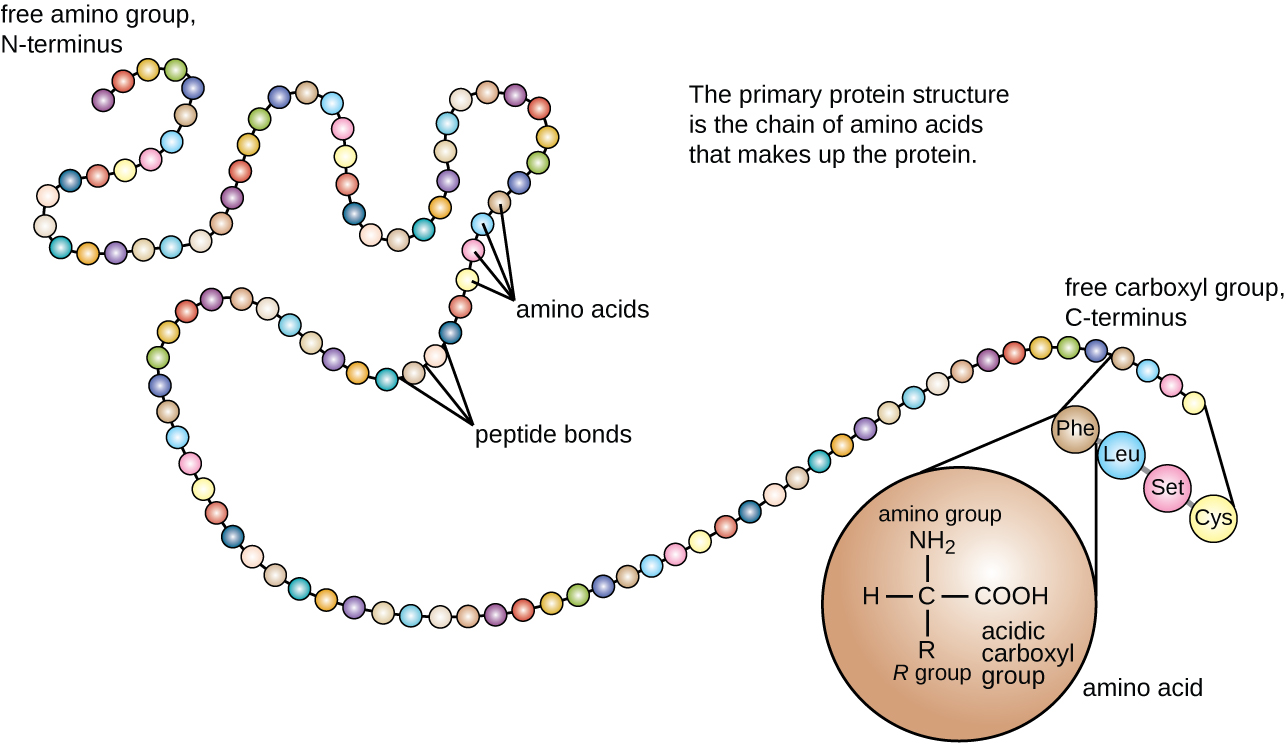
Protein sequencing is a foundational technique in molecular biology and biochemistry that involves determining the precise order of amino acids within a protein molecule. These amino acids are the building blocks of proteins and are arranged in a specific linear sequence, often referred to as the protein's primary structure. Understanding this primary structure is akin to deciphering the genetic code of proteins, and it holds immense significance across various scientific disciplines.
The Significance of Protein Sequencing
1. Decoding Genetic Information: At its core, protein sequencing serves as a bridge between the genetic information encoded in DNA and the functional proteins that carry out essential cellular processes. It allows scientists to translate the genetic code into the language of proteins, unveiling the specific sequence of amino acids that make up a protein.
2. Unveiling Protein Function: The primary structure of a protein is intimately linked to its function. Each protein's unique sequence dictates how it folds into its three-dimensional structure and, consequently, how it interacts with other molecules within a cell. Understanding this sequence is crucial for deciphering the roles proteins play in biological systems.
3. Implications in Biotechnology: In the field of biotechnology, protein sequencing is a fundamental step in the design and production of various biopharmaceuticals, enzymes, and genetically engineered proteins. By precisely sequencing target proteins, researchers can engineer these molecules for specific functions or therapeutic applications.
4. Personalized Medicine: In the context of medicine, protein sequencing plays a pivotal role in the emerging field of personalized medicine. It enables the identification of genetic mutations and variations that are associated with diseases, allowing for the development of tailored treatments and therapies for individuals.
5. Structural Biology Insights: Before researchers can delve into the three-dimensional structures of proteins (tertiary and quaternary structures), they must first determine the primary structure through protein sequencing. This information is essential for structural biologists seeking to understand how a protein's shape relates to its function and interactions.
6. Proteomics Advancements: In the broader context of proteomics, the study of all proteins within a biological system, protein sequencing is foundational. It facilitates the identification of proteins, their modifications, and their interactions, helping researchers unravel complex cellular processes and signaling pathways.
In essence, protein sequencing is the Rosetta Stone of the biological world, translating the genetic information stored in DNA into the functional language of proteins. It is a fundamental tool that underpins advances in biology, medicine, and biotechnology, empowering scientists to explore and harness the remarkable complexity of life at the molecular level.
Methods and Techniques for Protein Sequencing:
Edman Degradation
One of the earliest methods for protein sequencing is Edman degradation. This method selectively removes and identifies the N-terminal amino acid of a protein. While it was revolutionary in its time, Edman degradation has limitations, including the need for relatively large quantities of pure protein and challenges with repetitive sequences.
Mass Spectrometry
Mass spectrometry has emerged as a powerful tool for protein sequencing. It involves ionizing protein fragments and measuring their mass-to-charge ratios. Modern mass spectrometry techniques, such as tandem mass spectrometry (MS/MS), enable the identification of peptides and their sequences. This approach offers high sensitivity and can handle complex mixtures of proteins.
Next-Generation Sequencing
Recent advancements in next-generation sequencing (NGS) technologies have expanded their application beyond genomics to proteomics. NGS-based methods, like RNA-seq and ribosome profiling, can indirectly infer protein sequences by analyzing the corresponding mRNA sequences.
Each of these methods has its advantages and limitations. Edman degradation provides accurate results for small proteins but requires significant amounts of sample. Mass spectrometry is versatile and capable of analyzing complex mixtures but may struggle with membrane proteins. NGS offers high-throughput capabilities but relies on mRNA data and may not provide direct protein sequence information.
Details
Protein sequencing is the practical process of determining the amino acid sequence of all or part of a protein or peptide. This may serve to identify the protein or characterize its post-translational modifications. Typically, partial sequencing of a protein provides sufficient information (one or more sequence tags) to identify it with reference to databases of protein sequences derived from the conceptual translation of genes.
The two major direct methods of protein sequencing are mass spectrometry and Edman degradation using a protein sequenator (sequencer). Mass spectrometry methods are now the most widely used for protein sequencing and identification but Edman degradation remains a valuable tool for characterizing a protein's N-terminus.
Determining amino acid composition
It is often desirable to know the unordered amino acid composition of a protein prior to attempting to find the ordered sequence, as this knowledge can be used to facilitate the discovery of errors in the sequencing process or to distinguish between ambiguous results. Knowledge of the frequency of certain amino acids may also be used to choose which protease to use for digestion of the protein. The misincorporation of low levels of non-standard amino acids (e.g. norleucine) into proteins may also be determined. A generalized method often referred to as amino acid analysis[2] for determining amino acid frequency is as follows:
* Hydrolyse a known quantity of protein into its constituent amino acids.
* Separate and quantify the amino acids in some way.
Hydrolysis
Hydrolysis is done by heating a sample of the protein in 6 M hydrochloric acid to 100–110 °C for 24 hours or longer. Proteins with many bulky hydrophobic groups may require longer heating periods. However, these conditions are so vigorous that some amino acids (serine, threonine, tyrosine, tryptophan, glutamine, and cysteine) are degraded. To circumvent this problem, Biochemistry Online suggests heating separate samples for different times, analysing each resulting solution, and extrapolating back to zero hydrolysis time. Rastall suggests a variety of reagents to prevent or reduce degradation, such as thiol reagents or phenol to protect tryptophan and tyrosine from attack by chlorine, and pre-oxidising cysteine. He also suggests measuring the quantity of ammonia evolved to determine the extent of amide hydrolysis.
Separation and quantitation
The amino acids can be separated by ion-exchange chromatography then derivatized to facilitate their detection. More commonly, the amino acids are derivatized then resolved by reversed phase HPLC.
An example of the ion-exchange chromatography is given by the NTRC using sulfonated polystyrene as a matrix, adding the amino acids in acid solution and passing a buffer of steadily increasing pH through the column. Amino acids are eluted when the pH reaches their respective isoelectric points. Once the amino acids have been separated, their respective quantities are determined by adding a reagent that will form a coloured derivative. If the amounts of amino acids are in excess of 10 nmol, ninhydrin can be used for this; it gives a yellow colour when reacted with proline, and a vivid purple with other amino acids. The concentration of amino acid is proportional to the absorbance of the resulting solution. With very small quantities, down to 10 pmol, fluorescent derivatives can be formed using reagents such as ortho-phthaldehyde (OPA) or fluorescamine.
Pre-column derivatization may use the Edman reagent to produce a derivative that is detected by UV light. Greater sensitivity is achieved using a reagent that generates a fluorescent derivative. The derivatized amino acids are subjected to reversed phase chromatography, typically using a C8 or C18 silica column and an optimised elution gradient. The eluting amino acids are detected using a UV or fluorescence detector and the peak areas compared with those for derivatised standards in order to quantify each amino acid in the sample.
N-terminal amino acid analysis
Determining which amino acid forms the N-terminus of a peptide chain is useful for two reasons: to aid the ordering of individual peptide fragments' sequences into a whole chain, and because the first round of Edman degradation is often contaminated by impurities and therefore does not give an accurate determination of the N-terminal amino acid. A generalised method for N-terminal amino acid analysis follows:
* React the peptide with a reagent that will selectively label the terminal amino acid.
* Hydrolyse the protein.
* Determine the amino acid by chromatography and comparison with standards.
There are many different reagents which can be used to label terminal amino acids. They all react with amine groups and will therefore also bind to amine groups in the side chains of amino acids such as lysine - for this reason it is necessary to be careful in interpreting chromatograms to ensure that the right spot is chosen. Two of the more common reagents are Sanger's reagent (1-fluoro-2,4-dinitrobenzene) and dansyl derivatives such as dansyl chloride. Phenylisothiocyanate, the reagent for the Edman degradation, can also be used. The same questions apply here as in the determination of amino acid composition, with the exception that no stain is needed, as the reagents produce coloured derivatives and only qualitative analysis is required. So the amino acid does not have to be eluted from the chromatography column, just compared with a standard. Another consideration to take into account is that, since any amine groups will have reacted with the labelling reagent, ion exchange chromatography cannot be used, and thin-layer chromatography or high-pressure liquid chromatography should be used instead.
C-terminal amino acid analysis
The number of methods available for C-terminal amino acid analysis is much smaller than the number of available methods of N-terminal analysis. The most common method is to add carboxypeptidases to a solution of the protein, take samples at regular intervals, and determine the terminal amino acid by analysing a plot of amino acid concentrations against time. This method will be very useful in the case of polypeptides and protein-blocked N termini. C-terminal sequencing would greatly help in verifying the primary structures of proteins predicted from DNA sequences and to detect any posttranslational processing of gene products from known codon sequences.
It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
Pages: 1