Math Is Fun Forum
You are not logged in.
- Topics: Active | Unanswered
#1051 2021-06-15 00:24:09
- Jai Ganesh
- Administrator

- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
1029) Uncertainty principle
Uncertainty principle, also called Heisenberg uncertainty principle or indeterminacy principle, statement, articulated (1927) by the German physicist Werner Heisenberg, that the position and the velocity of an object cannot both be measured exactly, at the same time, even in theory. The very concepts of exact position and exact velocity together, in fact, have no meaning in nature.
Ordinary experience provides no clue of this principle. It is easy to measure both the position and the velocity of, say, an automobile, because the uncertainties implied by this principle for ordinary objects are too small to be observed. The complete rule stipulates that the product of the uncertainties in position and velocity is equal to or greater than a tiny physical quantity, or constant (h/(4π), where h is Planck’s constant, or about 6.6 × {10}^{-34} joule-second). Only for the exceedingly small masses of atoms and subatomic particles does the product of the uncertainties become significant.
Any attempt to measure precisely the velocity of a subatomic particle, such as an electron, will knock it about in an unpredictable way, so that a simultaneous measurement of its position has no validity. This result has nothing to do with inadequacies in the measuring instruments, the technique, or the observer; it arises out of the intimate connection in nature between particles and waves in the realm of subatomic dimensions.
The uncertainty principle arises from the wave-particle duality. Every particle has a wave associated with it; each particle actually exhibits wavelike behaviour. The particle is most likely to be found in those places where the undulations of the wave are greatest, or most intense. The more intense the undulations of the associated wave become, however, the more ill-defined becomes the wavelength, which in turn determines the momentum of the particle. So a strictly localized wave has an indeterminate wavelength; its associated particle, while having a definite position, has no certain velocity. A particle wave having a well-defined wavelength, on the other hand, is spread out; the associated particle, while having a rather precise velocity, may be almost anywhere. A quite accurate measurement of one observable involves a relatively large uncertainty in the measurement of the other.
The uncertainty principle is alternatively expressed in terms of a particle’s momentum and position. The momentum of a particle is equal to the product of its mass times its velocity. Thus, the product of the uncertainties in the momentum and the position of a particle equals h/(4π) or more. The principle applies to other related (conjugate) pairs of observables, such as energy and time: the product of the uncertainty in an energy measurement and the uncertainty in the time interval during which the measurement is made also equals h/(4π) or more. The same relation holds, for an unstable atom or nucleus, between the uncertainty in the quantity of energy radiated and the uncertainty in the lifetime of the unstable system as it makes a transition to a more stable state.

It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1052 2021-06-16 00:42:23
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
1030) Agoraphobia
Fear of Open or Crowded Spaces Phobia – Agoraphobia
Agoraphobia is the irrational fear of having a panic (or anxiety) attack in a place that may be difficult to escape from. Before we learn about the causes, symptoms and treatment of this phobia, let us first see a few myths associated with it and the actual facts.
Myths about Agoraphobia
• People with the fear of open spaces always remain housebound– Many sufferers of Agoraphobia actually prefer crowded spaces than being left alone at home. A majority of these patients may have milder symptoms of Agoraphobia. If one is housebound for months or years, then his/her Agoraphobia can be classified as being extreme.
• Agoraphobia is only the fear of crowded spaces– As mentioned above; some individuals are known to fear crowds while others to prefer them.
• Fear of enclosed spaces in not Agoraphobia, only claustrophobia (the fear of enclosed spaces)– Many individuals with Agoraphobia are also known to fear enclosed spaces but they might have other fear symptoms as well.
• Agoraphobia is the fear of open spaces and public places– More than the fear of being in an open space; the phobic tends to fear a “symptom-attack”- a rush of symptoms and sensations that s/he is unable to deal with.
• Agoraphobia is always a fear of panic attack– In Agoraphobia, it is not just ‘panic’ that one fears but several other symptoms. For example, a person might feel nauseated in a crowded space and fear not being able to reach the bathroom on time to throw up. Thus, the sufferer might “learn to feel or expect to feel something disturbing” in a particular situation and hence try to avoid the situations as much as possible.
Causes of Agoraphobia or the fear of open/crowded spaces
There is no single cause for the fear of open or crowded spaces. Researchers believe that a number of physical and psychological factors may be responsible for this phobia.
• In the majority of cases, an underlying ‘panic disorder’ may be responsible for Agoraphobia. A panic disorder is characterized by an intense and irrational fear that can cause the sufferer to lose control, cry, shake and have thoughts about dying. In his/her mind, the sufferer then links the attack to situations and then tries to avoid those situations completely.
• A research is also suggesting a possible link between long term tranquilizer or sleeping pill usage with Agoraphobia.
• Individuals with difficulty of spatial orientation and balance (or those with weaker vestibular functions) are also known to experience the extreme fear of crowded or open spaces.
• A history of alcohol or drug abuse, traumatic childhood experiences, recent life changes such as death, divorce, relationship difficulties, war, explosion, earthquakes etc can bring on the fear of open or crowded spaces.
Symptoms of Agoraphobia
The symptoms of this phobia can be classified into physical and psychological symptoms.
Physical symptoms:
• Hyperventilating or rapid/shallow breathing
• Feeling of choking or difficulty swallowing
• Sweating
• Shaking and trembling
• Nausea and other gastrointestinal distress
• Dizziness or lightheadedness
• Ringing or buzzing in the ears
Psychological symptoms
• Fear of losing control or going crazy
• Fear of dying
• Feeling ‘unreal’ or detached from oneself
• Feelings of depression, dread or anxiety
• Having low self esteem or low confidence
Treatment for Agoraphobia
It is essential to treat Agoraphobia early on, since, left untreated, it may take a more serious form and even make the sufferer depressed
There are several treatment options for dealing with the fear of open or crowded spaces. Of these, it is best to rely on the self help techniques rather than taking medications as the latter can have withdrawal symptoms and other side effects.
Self help techniques for dealing with panic symptoms
• Breathing slowly and counting to ten while repeating the word ‘relax’ in calm and soothing manner. This is one of the expert recommended self help techniques that have been proven highly effective in managing panic symptoms.
• Slowly exposing oneself to one’s fears and also writing down things that make one feels fearful. This might turn out to be difficult in the beginning but gradually one can overcome the fear of crowded or open spaces.
• Educating self – There are many books and case studies available online and offline that can inspire one to fight their Agoraphobia.
Other than these self help methods, one can also opt for CBT/cognitive behavior or behavior therapy, guided imagery, counseling, talk therapy and group therapy. Taking baby steps is the key to overcoming Agoraphobia.

It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1053 2021-06-17 00:18:32
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
1031) Boyle's law
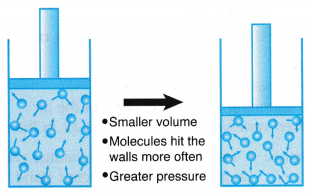
Boyle’s law, also called Mariotte’s law, a relation concerning the compression and expansion of a gas at constant temperature. This empirical relation, formulated by the physicist Robert Boyle in 1662, states that the pressure (p) of a given quantity of gas varies inversely with its volume (v) at constant temperature; i.e., in equation form, pv = k, a constant. The relationship was also discovered by the French physicist Edme Mariotte (1676).
The law can be derived from the kinetic theory of gases assuming a perfect (ideal) gas. Real gases obey Boyle’s law at sufficiently low pressures, although the product pv generally decreases slightly at higher pressures, where the gas begins to depart from ideal behaviour.
Boyle’s Law is a basic law in chemistry describing the behavior of a gas held at a constant temperature. The law, discovered by Robert A. Boyle in 1662, states that at a fixed temperature, the volume of gas is inversely proportional to the pressure exerted by the gas. In other words, when a gas is pumped into an enclosed space, it will shrink to fit into that space, but the pressure that gas puts on the container will increase.
Perhaps a more straightforward way is to say Boyle's law is the relationship between pressure and volume. Mathematically, Boyle’s law can be written as pV=k, where p is the pressure of the gas, V is the volume of the gas, and k is a constant.
An example of Boyle’s law in action can be seen in a balloon. Air is blown into the balloon; the pressure of that air pushes on the rubber, making the balloon expand. If one end of the balloon is squeezed, making the volume smaller, the pressure inside increased, making the un-squeezed part of the balloon expand out. There is a limit to how much the air/gas can be compressed, however, because eventually the pressure becomes so great that it causes the balloon to break.
It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1054 2021-06-18 00:04:53
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
1032) Charles's law
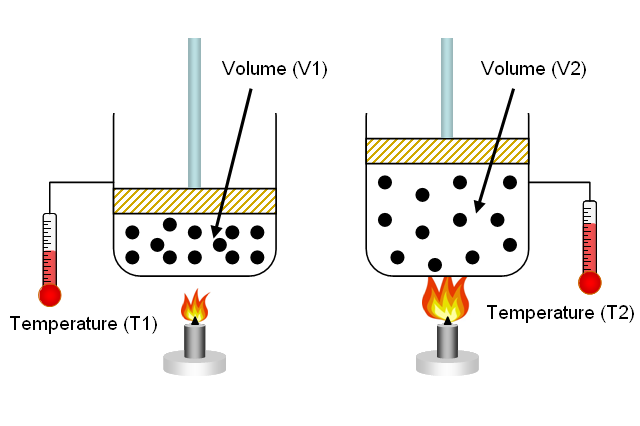
Charles’s law, a statement that the volume occupied by a fixed amount of gas is directly proportional to its absolute temperature, if the pressure remains constant. This empirical relation was first suggested by the French physicist J.-A.-C. Charles about 1787 and was later placed on a sound empirical footing by the chemist Joseph-Louis Gay-Lussac. It is a special case of the general gas law and can be derived from the kinetic theory of gases under the assumption of a perfect (ideal) gas. Measurements show that at constant pressure the thermal expansion of real gases, at sufficiently low pressure and high temperature, conforms closely to Charles’s law.
The physical principle known as Charles' law states that the volume of a gas equals a constant value multiplied by its temperature as measured on the Kelvin scale (zero Kelvin corresponds to -273.15 degrees Celsius).
Charles's law, a statement that the volume occupied by a fixed amount of gas is directly proportional to its absolute temperature, if the pressure remains constant. It is a special case of the general gas law and can be derived from the kinetic theory of gases under the assumption of a perfect (ideal) gas.
It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1055 2021-06-19 00:07:48
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
1033) Ohm's law
Ohm’s law, description of the relationship between current, voltage, and resistance. The amount of steady current through a large number of materials is directly proportional to the potential difference, or voltage, across the materials. Thus, if the voltage V (in units of volts) between two ends of a wire made from one of these materials is tripled, the current I (amperes) also triples; and the quotient V/I remains constant. The quotient V/I for a given piece of material is called its resistance, R, measured in units named ohms. The resistance of materials for which Ohm’s law is valid does not change over enormous ranges of voltage and current. Ohm’s law may be expressed mathematically as V/I = R. That the resistance, or the ratio of voltage to current, for all or part of an electric circuit at a fixed temperature is generally constant had been established by 1827 as a result of the investigations of the German physicist Georg Simon Ohm.
Alternate statements of Ohm’s law are that the current I in a conductor equals the potential difference V across the conductor divided by the resistance of the conductor, or simply I = V/R, and that the potential difference across a conductor equals the product of the current in the conductor and its resistance, V = IR. In a circuit in which the potential difference, or voltage, is constant, the current may be decreased by adding more resistance or increased by removing some resistance. Ohm’s law may also be expressed in terms of the electromotive force, or voltage, E, of the source of electric energy, such as a battery. For example, I = E/R.
With modifications, Ohm’s law also applies to alternating-current circuits, in which the relation between the voltage and the current is more complicated than for direct currents. Precisely because the current is varying, besides resistance, other forms of opposition to the current arise, called reactance. The combination of resistance and reactance is called impedance, Z. When the impedance, equivalent to the ratio of voltage to current, in an alternating current circuit is constant, a common occurrence, Ohm’s law is applicable. For example, V/I = Z.
With further modifications Ohm’s law has been extended to the constant ratio of the magnetomotive force to the magnetic flux in a magnetic circuit.

It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1056 2021-06-20 00:04:30
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
1034) Coulomb's law
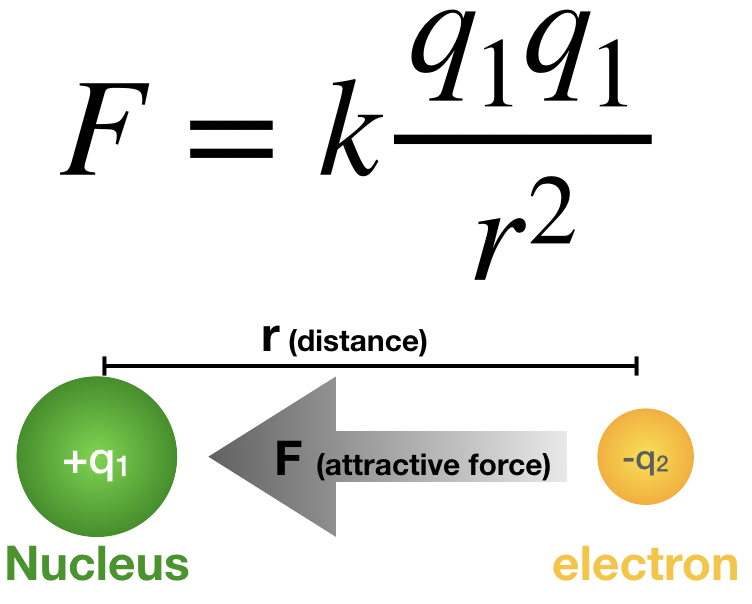
Coulomb’s law, mathematical description of the electric force between charged objects. Formulated by the 18th-century French physicist Charles-Augustin de Coulomb, it is analogous to Isaac Newton’s law of gravity.
Both gravitational and electric forces decrease with the square of the distance between the objects, and both forces act along a line between them. In Coulomb’s law, however, the magnitude and sign of the electric force are determined by the electric charge, rather than the mass, of an object. Thus, charge determines how electromagnetism influences the motion of charged objects. Charge is a basic property of matter. Every constituent of matter has an electric charge with a value that can be positive, negative, or zero. For example, electrons are negatively charged, and atomic nuclei are positively charged. Most bulk matter has an equal amount of positive and negative charge and thus has zero net charge.
According to Coulomb, the electric force for charges at rest has the following properties:
1. Like charges repel each other; unlike charges attract. Thus, two negative charges repel one another, while a positive charge attracts a negative charge.
2. The attraction or repulsion acts along the line between the two charges.
3. The size of the force varies inversely as the square of the distance between the two charges. Therefore, if the distance between the two charges is doubled, the attraction or repulsion becomes weaker, decreasing to one-fourth of the original value. If the charges come 10 times closer, the size of the force increases by a factor of 100.
4. The size of the force is proportional to the value of each charge. The unit used to measure charge is the coulomb (C). If there were two positive charges, one of 0.1 coulomb and the second of 0.2 coulomb, they would repel each other with a force that depends on the product 0.2 × 0.1. Thus, if each of the charges were reduced by one-half, the repulsion would be reduced to one-quarter of its former value.
It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1057 2021-06-21 00:08:33
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
1035) Ampère's law
Ampère’s law, one of the basic relations between electricity and magnetism, stating quantitatively the relation of a magnetic field to the electric current or changing electric field that produces it. The law is named in honour of André-Marie Ampère, who by 1825 had laid the foundation of electromagnetic theory. An alternative expression of the Biot-Savart law (q.v.), which also relates the magnetic field and the current that produces it, Ampère’s law is generally stated formally in the language of calculus: the line integral of the magnetic field around an arbitrarily chosen path is proportional to the net electric current enclosed by the path. James Clerk Maxwell is responsible for this mathematical formulation and for the extension of the law to include magnetic fields that arise without electric current, as between the plates of a capacitor, or condenser, in which the electric field changes with the periodic charging and discharging of the plates but in which no passage of electric charge occurs. Maxwell also showed that even in empty space a varying electric field is accompanied by a changing magnetic field. In this more general form, the so-called Ampère-Maxwell law is one of the four Maxwell equations that define electromagnetism.
Ampere's Law can be stated as:
“The magnetic field created by an electric current is proportional to the size of that electric current with a constant of proportionality equal to the permeability of free space.”

It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1058 2021-06-22 00:07:22
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
1036) Newton's law of cooling
Newton's law of cooling states that ‘the rate of heat loss of a body is directly proportional to the difference in the temperatures between the body and its surroundings’. The law is frequently qualified to include the condition that the temperature difference is small and the nature of heat transfer mechanism remains the same. As such, it is equivalent to a statement that the heat transfer coefficient, which mediates between heat losses and temperature differences, is a constant. This condition is generally met in heat conduction (where it is guaranteed by Fourier's law) as the thermal conductivity of most materials is only weakly dependent on temperature. In convective heat transfer, Newton's Law is followed for forced air or pumped fluid cooling, where the properties of the fluid do not vary strongly with temperature, but it is only approximately true for buoyancy-driven convection, where the velocity of the flow increases with temperature difference. Finally, in the case of heat transfer by thermal radiation, Newton's law of cooling holds only for very small temperature differences, and a more accurate description is given by Planck's Law.
Sir Isaac Newton did not originally state his law in the above form in 1701, when it was originally formulated. Rather, using today's terms, Newton noted after some mathematical manipulation that ‘the rate of temperature change’ of a body is proportional to the difference in temperatures between the body and its surroundings. This final simplest version of the law given by Newton himself, was partly due to confusion in Newton's time between the concepts of heat and temperature, which would not be fully disentangled until much later.
When stated in terms of temperature differences, Newton's law (with several further simplifying assumptions, such as a low Biot number and a temperature-independent heat capacity) results in a simple differential equation temperature-difference as a function of time. The solution to that equation describes an exponential decrease of temperature-difference over time. This characteristic decay of the temperature-difference is also associated with Newton's law of cooling.
Relationship to mechanism of cooling
Convection cooling is sometimes said to be governed by "Newton's law of cooling." This use is based on a work by Sir Isaac Newton published anonymously in 1701 as "Scala graduum Caloris. Calorum Descriptiones & signa." in Philosophical Transactions, volume 22, issue 270.
When the heat transfer coefficient is independent, or relatively independent, of the temperature difference between object and environment, Newton's law is followed. This independence is sometimes the case, but is not generally so. The law holds well for forced air and pumped liquid cooling, where the fluid velocity does not rise with increasing temperature difference. Newton's law is most closely obeyed in purely conduction-type cooling. However, the heat transfer coefficient is a function of the temperature difference in natural convective (buoyancy driven) heat transfer. In that case, Newton's law only approximates the result when the temperature difference is relatively small. Newton himself realized this limitation.
A correction to Newton's law concerning convection for larger temperature differentials by including an exponent, was made in 1817 by Dulong and Petit. (These men are better-known for their formulation of the Dulong–Petit law concerning the molar specific heat capacity of a crystal.)
Another situation which also does not obey Newton's law, is radiative heat transfer, being better described by Planck's law as varying with the 4th power of absolute temperature.

It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1059 2021-06-23 00:20:32
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
1037) Hooke's law
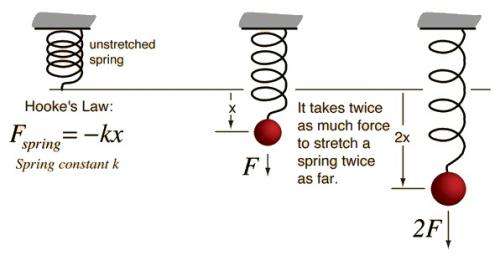
Hooke’s law, law of elasticity discovered by the English scientist Robert Hooke in 1660, which states that, for relatively small deformations of an object, the displacement or size of the deformation is directly proportional to the deforming force or load. Under these conditions the object returns to its original shape and size upon removal of the load. Elastic behaviour of solids according to Hooke’s law can be explained by the fact that small displacements of their constituent molecules, atoms, or ions from normal positions is also proportional to the force that causes the displacement.
The deforming force may be applied to a solid by stretching, compressing, squeezing, bending, or twisting. Thus, a metal wire exhibits elastic behaviour according to Hooke’s law because the small increase in its length when stretched by an applied force doubles each time the force is doubled. Mathematically, Hooke’s law states that the applied force F equals a constant k times the displacement or change in length x, or F = kx. The value of k depends not only on the kind of elastic material under consideration but also on its dimensions and shape.
At relatively large values of applied force, the deformation of the elastic material is often larger than expected on the basis of Hooke’s law, even though the material remains elastic and returns to its original shape and size after removal of the force. Hooke’s law describes the elastic properties of materials only in the range in which the force and displacement are proportional. Sometimes Hooke’s law is formulated as F = −kx. In this expression F no longer means the applied force but rather means the equal and oppositely directed restoring force that causes elastic materials to return to their original dimensions.
Hooke’s law may also be expressed in terms of stress and strain. Stress is the force on unit areas within a material that develops as a result of the externally applied force. Strain is the relative deformation produced by stress. For relatively small stresses, stress is proportional to strain.
It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1060 2021-06-24 00:19:13
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
1038) Snell's law
Snell’s law, in optics, a relationship between the path taken by a ray of light in crossing the boundary or surface of separation between two contacting substances and the refractive index of each. This law was discovered in 1621 by the Dutch astronomer and mathematician Willebrord Snell (also called Snellius). The account of Snell’s law went unpublished until its mention by Christiaan Huygens in his treatise on light. n1 and n2 represent the indices of refraction for the two media, and α1 and α2 are the angles of incidence and refraction that the ray R makes with the normal (perpendicular) line NN at the boundary. Snell’s law asserts that n1/n2 = sin α2/sin α1.
Because the ratio n1/n2 is a constant for any given wavelength of light, the ratio of the two sines is also a constant for any angle. Thus, the path of a light ray is bent toward the normal when the ray enters a substance with an index of refraction higher than the one from which it emerges; and because the path of a ray of light is reversible, the ray is bent away from the normal when entering a substance of lower refractive index.
The reason light is refracted in going from one medium to another.

It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1061 2021-06-25 00:13:29
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
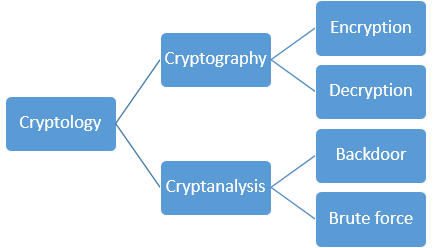
1039) Cryptography and Cryptology
Cryptography, Practice of the enciphering and deciphering of messages in secret code in order to render them unintelligible to all but the intended receiver. Cryptography may also refer to the art of cryptanalysis, by which cryptographic codes are broken. Collectively, the science of secure and secret communications, involving both cryptography and cryptanalysis, is known as cryptology. The principles of cryptography are today applied to the encryption of fax, television, and computer network communications. In particular, the secure exchange of computer data is of great importance to banking, government, and commercial communications.
Cryptology, science concerned with data communication and storage in secure and usually secret form. It encompasses both cryptography and cryptanalysis.
The term cryptology is derived from the Greek kryptós (“hidden”) and lógos (“word”). Security obtains from legitimate users being able to transform information by virtue of a secret key or keys—i.e., information known only to them. The resulting cipher, although generally inscrutable and not forgeable without the secret key, can be decrypted by anyone knowing the key either to recover the hidden information or to authenticate the source. Secrecy, though still an important function in cryptology, is often no longer the main purpose of using a transformation, and the resulting transformation may be only loosely considered a cipher.
Cryptography (from the Greek kryptós and gráphein, “to write”) was originally the study of the principles and techniques by which information could be concealed in ciphers and later revealed by legitimate users employing the secret key. It now encompasses the whole area of key-controlled transformations of information into forms that are either impossible or computationally infeasible for unauthorized persons to duplicate or undo.
Cryptanalysis (from the Greek kryptós and analýein, “to loosen” or “to untie”) is the science (and art) of recovering or forging cryptographically secured information without knowledge of the key. Cryptology is often—and mistakenly—considered a synonym for cryptography and occasionally for cryptanalysis, but specialists in the field have for years adopted the convention that cryptology is the more inclusive term, encompassing both cryptography and cryptanalysis.
Cryptography was initially only concerned with providing secrecy for written messages, especially in times of war. Its principles apply equally well, however, to securing data flowing between computers or data stored in them, to encrypting facsimile and television signals, to verifying the identity of participants in electronic commerce (e-commerce) and providing legally acceptable records of those transactions. Because of this broadened interpretation of cryptography, the field of cryptanalysis has also been enlarged.
It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1062 2021-06-26 00:30:46
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
1040) Salt
Salt (NaCl), sodium chloride, mineral substance of great importance to human and animal health, as well as to industry. The mineral form halite, or rock salt, is sometimes called common salt to distinguish it from a class of chemical compounds called salts.
Salt is essential to the health of both people and animals. Table salt, used universally as a seasoning, is fine-grained and of high purity. To ensure that this hygroscopic (i.e., water-attracting) substance will remain free-flowing when exposed to the atmosphere, small quantities of sodium aluminosilicate, tricalcium phosphate, or magnesium silicate are added. Iodized salt—that is, salt to which small quantities of potassium iodide have been added—is widely used in areas where iodine is lacking from the diet, a deficiency that can cause swelling of the thyroid gland, commonly called goitre. Livestock also require salt; it is often made available in solid blocks.
The meat-packing, sausage-making, fish-curing, and food-processing industries use salt as a preservative or seasoning or both. It is employed for curing and preserving hides and as a brine for refrigeration.
In the chemical industry, salt is required in the manufacture of sodium bicarbonate (baking soda), sodium hydroxide (caustic soda), hydrochloric acid, chlorine, and many other chemicals. Salt is also employed in soap, glaze, and porcelain enamel manufacture and enters into metallurgical processes as a flux (a substance promoting fusing of metals).
When applied to snow or ice, salt lowers the melting point of the mixture. Thus, large amounts are used in northern climates to help rid thoroughfares of accumulated snow and ice. Salt is used in water-softening equipment that removes calcium and magnesium compounds from water.
History Of Use
In some parts of the Western Hemisphere and in India, the use of salt was introduced by Europeans, but in parts of central Africa it is still a luxury available only to the rich. Where people live mainly on milk and raw or roasted meat (so that its natural salts are not lost), sodium chloride supplements are unnecessary; nomads with their flocks of sheep or herds of cattle, for example, never eat salt with their food. On the other hand, people who live mostly on cereal, vegetable, or boiled meat diets require supplements of salt.
The habitual use of salt is intimately connected with the advance from nomadic to agricultural life, a step in civilization that profoundly influenced the rituals and cults of almost all ancient nations. The gods were worshipped as the givers of the kindly fruits of the earth, and salt was usually included in sacrificial offerings consisting wholly or partly of cereal elements. Such offerings were prevalent among the Greeks and Romans and among a number of the Semitic peoples.
Covenants were ordinarily made over a sacrificial meal, in which salt was a necessary element. The preservative qualities of salt made it a peculiarly fitting symbol of an enduring compact, sealing it with an obligation to fidelity. The word salt thus acquired connotations of high esteem and honour in ancient and modern languages. Examples include the Arab avowal “There is salt between us,” the Hebrew expression “to eat the salt of the palace,” and the modern Persian phrase namak ḥarām, “untrue to salt” (i.e., disloyal or ungrateful). In English the term “salt of the earth” describes a person held in high esteem.
Salt contributes greatly to our knowledge of the ancient highways of commerce. One of the oldest roads in Italy is the Via Salaria (Salt Route) over which Roman salt from Ostia was carried into other parts of Italy. Herodotus tells of a caravan route that united the salt oases of the Libyan Desert. The ancient trade between the Aegean and the Black Sea coast of southern Russia was largely dependent on the salt pans (ponds for evaporating seawater to obtain salt) at the mouth of the Dnieper River and on the salt fish brought from this district.
Cakes of salt have been used as money in Ethiopia and elsewhere in Africa and in Tibet. In the Roman army an allowance of salt was made to officers and men; in imperial times, this salarium (from which the English word salary is derived) was converted into an allowance of money for salt.
China, the United States, India, Germany, Canada, and Australia are the world’s largest salt producers in the early 21st century.
Occurrence
Seawater
Though the material that gives seawater its salty flavour is composed of many substances, sodium chloride, or common salt, is by far the predominant compound. On the assumption that 1 gallon (about 4 litres) of seawater contains 0.231 pound (about 105 grams) of salt and that rock salt on the average is 2.17 times as dense as water, it has been estimated that if the oceans of the world were completely dried up, they would yield at least 4.5 million cubic miles of rock salt, or about 14.5 times the bulk of the entire continent of Europe above the high-water mark.
Seawater contains on the average about 3 percent salt, although the actual concentration varies from about 1 percent (in the polar seas) to 5 percent. Enclosed waters such as the Mediterranean and Red seas contain a higher proportion of salt than does the open ocean at the same latitude. Irrespective of the source of the seawater, salt obtained by the evaporation of seawater has the following composition: sodium chloride 77.76 percent, magnesium chloride 10.88 percent, magnesium sulfate 4.74 percent, calcium sulfate 3.60 percent, potassium chloride 2.46 percent, magnesium bromide 0.22 percent, and calcium carbonate 0.34 percent.
Natural brines
Brine is water containing a high concentration of salt. Natural brines of commercial importance are found in the Dead Sea as well as in Austria, France, Germany, India, the United States, and the United Kingdom. Salt in brines is nearly always accompanied by chlorides and sulfates of potassium, calcium, and magnesium; carbonates and the element bromine often are present as well.
The Dead Sea, which covers an area of 1,020 square km (394 square miles), contains approximately 12,650,000,000 tons of salt. The Jordan River, which contains only 35 parts of salt per 100,000 parts of water, adds 850,000 tons of salt to this total each year.
The concentration of salts in the Dead Sea varies from 270 to 300 parts per thousand to a depth of 40 metres (130 feet); it increases gradually from 40 to 100 metres (130 to 330) feet and remains a fairly constant 332 parts per thousand below 100 metres. Dead Sea water is relatively free from sulfates and has a high proportion of potassium and bromine. Because atmospheric conditions favour evaporation by sunlight (solar evaporation) for about eight months of the year, the production of salt, potassium, and bromine is feasible in the Dead Sea area. The process used for recovery of salt and potash is similar to that described below under Salt manufacture. The Indian brines at Khārāghoda resemble seawater in the character of their dissolved salts but are much more concentrated and in some cases virtually saturated; that is, they have dissolved all the salt they can.
Certain natural brines occurring in the United Kingdom and the United States are of special interest because they contain salts, such as the chlorides of barium and strontium, that are not usually found in brines. Special processing methods are required to produce salt from such brines. In Britain these unusual brines are found at great depths during test drillings for petroleum, while in the United States such brines occur in deep wells in several places.
Rock salt
Rock salt is crystalline sodium chloride, called halite by mineralogists. It occurs widely in the form of rock masses and beds and is abundant in rocks from all geologic periods. Because of its great solubility in water, it occurs under extremely thick cover in humid regions but lies close to the surface in arid regions.
All major rock salt deposits originated from the evaporation of seawater at some time during the geologic past. Approximately 78 percent of the mineral matter in normal seawater is sodium chloride. Upon evaporation of about nine-tenths of the volume of seawater, rock salt is precipitated. Calcium sulfate (gypsum and anhydrite) and potassium and magnesium salts also are precipitated. Deposits are found in beds from a few feet to many hundreds of feet thick. The ages of these beds range through much of geologic time. Because evaporation of a large quantity of seawater leaves only a small amount of salt, it is theorized that many extremely thick rock salt beds were deposited in partly enclosed arms of the seas in which evaporation was greater than the inflow of salt water. A barrier on the seafloor at the entrance to the basin prevented the outflow of the concentrated saline water.
Such bedded salt deposits occur in the Punjab Salt Range in Pakistan and in Iran; however, these deposits have been little exploited. Similar deposits in the United States and Canada are worked extensively for both industrial and domestic use. Other important salt deposits, usually classified by the age of the surrounding rock, are found in Germany, Nova Scotia, the sub-Carpathian region extending from Poland through Hungary and Romania, and the province of Sichuan in China, where salt wells have been in existence for more than 2,000 years.
Another economically important type of rock salt deposit is the salt domes, which were formed when earth pressure forced up plugs of rock salt measuring approximately a mile across. The domes appear to result from pressure, which pushes the salt up through the rocks from depths as great as 50,000 feet (15,000 metres). Many domes occur at shallow depths and are extensively mined. Domes in the sub-Carpathian region of Europe have been worked since ancient times. The North German Plain has many extensively mined domes, which are thought to have originated below 6,000 feet; domes also are abundant along the U.S. Gulf Coast. Rock salt may be obtained from domes by the usual mining methods or by drilling wells into the salt strata and pumping water down to dissolve the salt; the brine is then returned to the surface, where it is processed like natural brine.
Salt Manufacture
At one time almost all the salt used in commerce was produced from the evaporation of seawater, and sea salt still is a staple commodity in many maritime countries, especially where the climate is dry and the summer is long. Commercial salt is manufactured from rock salt, as well as from seawater and other natural and artificial brines. Most of the artificial brines are obtained by pumping water into underground salt beds. A considerable amount of brine itself is used directly in industrial countries.
Manufacture from rock salt
The beds of rock salt are mined or quarried by the usual excavation methods, depending on the depths and thicknesses of the deposits and on local conditions. The mined rock salt sometimes is dissolved and the salt manufactured by treatment of the brine, as described below. The method affords opportunities for purification of the salt. When the rock salt is of a high degree of purity, as in Poland and the United States, the salt may be ground, screened, and marketed without further processing. The salt is mined in large lumps that are first crushed, then more finely ground and screened by size into various grades; the salt is then bulk-loaded into trucks, hoppers, or barges or loaded into bags for further handling. Bulk handling has been greatly facilitated by the use of anticaking agents which allow the salt to be stored uncovered and outdoors without becoming a hard mass again.
Manufacture from seawater and brines
Only a certain quantity of salt will dissolve in water at any given temperature. Once the solution contains as much salt as it can hold, it is said to be saturated; any further additions of salt will not dissolve.
Evaporation is the reverse of this process. When an aqueous solution of several salts (seawater, for example) is evaporated, each of the salts precipitates as it reaches its point of saturation in the solution. Thus, the different salts in seawater will precipitate at different times, forming layers on the bottom of the evaporating pond. For seawater and many brines, the order of deposition is calcium carbonate, calcium sulfate, sodium chloride, magnesium sulfate, potassium magnesium chloride, and magnesium chloride.
Solar evaporation
In maritime countries where there is a negative evaporation rate—i.e., the amount of water evaporating exceeds the amount of rainfall by at least 75 cm (about 30 inches)—salt is produced by solar evaporation from seawater. The processes used are similar in general principle from country to country, but details of equipment vary from sophisticated in the United States to quite primitive in developing nations.
A preliminary concentration is usually accomplished by allowing the seawater to flow through a series of gates constructed of wood or a combination of wood and concrete into a series of shallow ponds separated by dikes. In these ponds the solution is concentrated to a specific gravity of about 1.22; this means that a given volume of brine is 1.22 times as dense as a given volume of pure water. At this stage, suspended impurities such as sand, clay, and the less soluble salts such as calcium carbonate, or chalk, and calcium sulfate are removed. Solar evaporation of the Dead Sea water is hastened by adding dye to the water. The dye permits more heat to be absorbed from sunlight in thinner layers of brine so that shallow ponds may be used and the penetration of brine into the ground is reduced.
Once it has been concentrated, the brine is run through a series of crystallizing pans, usually four in number, where the salt is deposited as evaporation proceeds. In the first crystallizing pan, the brine is concentrated to a specific gravity of 1.23 and remains partly contaminated with calcium sulfate. The specific gravity of the solution in the pan increases slowly during crystallization of the salt, reaching 1.24 in the second pan. In the third pan the specific gravity of the solution reaches 1.25, and the salt deposited there contains small amounts of magnesium sulfate as an impurity. The final solution, termed bitterns, has a specific gravity of 1.25–1.26 and is used in some countries (United States and Israel) in the manufacture of potash, bromine, epsom salts (magnesium sulfate), and magnesium chloride.
In developing countries the salt in each crystallizing pan is raked into rows, where it is allowed to drain for several days. After that it is collected into heaps, drained again, lifted from the pans, and finally dried. In industrial countries the salt is harvested mechanically and washed with saturated brine. It is then dewatered, washed with fresh water, and stored for further processing or direct sale.
Use of artificial heat
In areas where bedded deposits can be solution-mined, evaporated salt is recovered from these solutions with artificial heat. Some evaporated salt also is made from natural brine or solar salt. Formerly, brine was concentrated in open pans over fire. More recently, steam-jacketed vessels have been used. The largest amount of salt produced in the colder climates is rock salt. The largest amount of evaporated salt is produced by multiple-effect vacuum evaporators, and an important quantity is made in so-called open crystallizers or grainers that produce a type of crystal preferred for use in some of the food industries. The brine, natural or artificial, is first pumped into settling tanks, where calcium and magnesium compounds may be removed by chemical treatment. In grainer operations the settled and filtered brine is delivered to the grainer, a long open trough heated with steam coils. The brine is fed into the grainer at approximately the same rate at which evaporation is taking place and at a temperature only slightly below that of the brine in the grainer. The residue of brine, or bitterns, may be removed continuously, once a day, or less often. Evaporation occurs at the surface of the liquid, and the crystals originate there. They remain at the surface, held up by the surface tension of the brine. The crystal grows at the top edges, becoming a small inverted hollow pyramid, or hopper. Eventually the hopper sinks and ceases to grow. When the crystals are recovered, the salt is largely in the form of flakes, hence the name flake salt.
When multiple-effect evaporators are used, the vacuum in each vessel is adjusted so that the vapour from the first vessel is hot enough to boil the brine in the second, the vapour from the second supplying the heat to operate the third vessel or effect. The brine is usually sent through the stages or effects in succession, although in the case of salt manufacture fresh brine may be fed to each stage if desired. With open pans, 4,500 to 5,400 kg (10,000 to 12,000 pounds) of steam are required to produce 900 kg (1 ton) of salt. With triple-effect evaporation, 630 kg (1,400 pounds) of steam produce 1 ton of salt.
The Alberger process is partially a vacuum-pan and partially a grainer operation in which cubic crystals are formed in the solution fed to the grainer pans by a partial vacuum-pan evaporation. These seed crystals in the grainer produce a salt that is a mixture of the grainer-type flake and the flake grown on seed crystals. About 1,360 kg (3,000 pounds) of steam are required to produce one ton of salt. Salt from the Alberger process is centrifuged (spun) from the brine and then dried. Table salt may have small amounts of aluminum calcium silicate, calcium silicate, magnesium silicate, tricalcium silicate, magnesium carbonate, or tricalcium phosphate added to keep it free-flowing. Iodized salt has potassium iodide added. In some countries yellow prussiate of soda, to prevent caking, is added in minute amounts as regulated by the government.

It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1063 2021-06-27 00:27:01
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
1041) Artesian well
Artesian well, well from which water flows under natural pressure without pumping. It is dug or drilled wherever a gently dipping, permeable rock layer (such as sandstone) receives water along its outcrop at a level higher than the level of the surface of the ground at the well site. At the outcrop the water moves down into the aquifer (water-bearing layer) but is prevented from leaving it, by impermeable rock layers (such as shale) above and below it. Pressure from the water’s weight (hydrostatic pressure) forces water to the surface of a well drilled down into the aquifer; the pressure for the steady upflow is maintained by the continuing penetration of water into the aquifer at the intake area.
In places where the overlying impermeable rocks are broken by joints or faults, water may escape through them to rise to the surface as artesian springs. In some areas, artesian wells and springs are a major source of water, especially in arid plains adjacent to mountain ranges that receive precipitation. The rapid development of new wells through over-drilling, however, has tended to reduce head pressures in many artesian systems. As a result, most artesian wells are now outfitted with pumps.
An artesian well may sound like a fancy place to get your water, but it’s really nothing more than a well tapping groundwater that is under pressure. In some artesian wells, the pressure is high enough that a pump isn’t needed to bring water to the surface. These wells are known as flowing artesian wells.
Artesian wells occur naturally over large areas of the Santa Clara Valley, and flowing artesian wells have been observed since the 1850s. When pressures in groundwater aquifers are high, old lost wells may begin flowing freely at the land surface. This wastes water and can create nuisance conditions. Recent wet winters and low groundwater pumping are causing higher pressures in artesian aquifers. This is causing more old wells to start flowing and leading to more frequent discoveries of old abandoned wells.
Whether they are flowing or not, the problem with abandoned wells is they can provide a way for contaminants to move into groundwater or between groundwater aquifers. That’s why they’re not allowed by state law or Valley Water ordinance.
Groundwater is a key source of water for Santa Clara County – our groundwater basins can hold more water than all 10 of our surface water reservoirs combined – and Valley Water works hard to protect the quality of this water that is so important to life in our county.
All wells that are no longer in use must be properly destroyed to avoid groundwater contamination. Artesian wells are the responsibility of the property owner where the well is found.
If you see a lot of water on the ground, how do you know if it’s coming from an artesian well?
Things to look for:
(a) Water at the land surface with no known source: it’s not leaking from pipes; it’s not from shallow groundwater (usually found in low spots on freeways, during excavation or in basements, etc.); and it’s not from a spring (typically found on or at the base of a hill slope);
(b) There may be bubbles in the water;
(c) You actually see water emanating from a well.

It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1064 2021-06-28 00:14:21
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
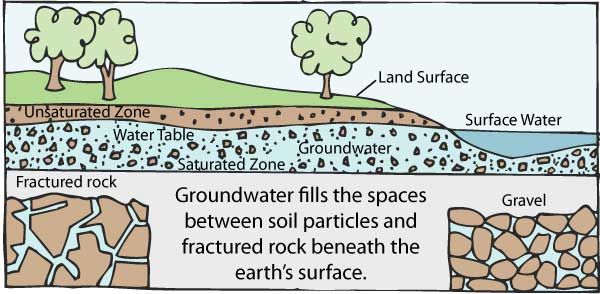
1042) Groundwater
Groundwater, water that occurs below the surface of Earth, where it occupies all or part of the void spaces in soils or geologic strata. It is also called subsurface water to distinguish it from surface water, which is found in large bodies like the oceans or lakes or which flows overland in streams. Both surface and subsurface water are related through the hydrologic cycle (the continuous circulation of water in the Earth-atmosphere system).
A brief treatment of groundwater follows.
Most groundwater comes from precipitation. Precipitation infiltrates below the ground surface into the soil zone. When the soil zone becomes saturated, water percolates downward. A zone of saturation occurs where all the interstices are filled with water. There is also a zone of aeration where the interstices are occupied partially by water and partially by air. Groundwater continues to descend until, at some depth, it merges into a zone of dense rock. Water is contained in the pores of such rocks, but the pores are not connected and water will not migrate. The process of precipitation replenishing the groundwater supply is known as recharge. In general, recharge occurs only during the rainy season in tropical climates or during winter in temperate climates. Typically, 10 to 20 percent of the precipitation that falls to the Earth enters water-bearing strata, which are known as aquifers.
Groundwater is constantly in motion. Compared to surface water, it moves very slowly, the actual rate dependent on the transmissivity and storage capacity of the aquifer. Natural outflows of groundwater take place through springs and riverbeds when the groundwater pressure is higher than atmospheric pressure in the vicinity of the ground surface. Internal circulation is not easily determined, but near the water table the average cycling time of water may be a year or less, while in deep aquifers it may be as long as thousands of years.
Groundwater plays a vital role in the development of arid and semiarid zones, sometimes supporting vast agricultural and industrial enterprises that could not otherwise exist. It is particularly fortunate that aquifers antedating the formation of deserts remain unaffected by increases in aridity with the passage of time. Withdrawal, however, will deplete even the largest of groundwater basins so that development based on the existence of aquifers can be only temporary at best.
A vast amount of groundwater is distributed throughout the world, and a large number of groundwater reservoirs are still underdeveloped or uninvestigated. Scientists estimate that some 5.97 quintillion gallons (22.6 million cubic km [5.4 million cubic miles]) of groundwater reside in the upper 2 km (1.2 miles) of Earth’s surface. The most frequently investigated or exploited groundwater reservoirs are of the unconsolidated clastic (mainly sand and gravel) or carbonate hardrock type found in alluvial valleys and coastal plains under temperate or arid conditions.
Though some groundwater dissolves substances from rocks and may contain traces of old seawater, most groundwater is free of pathogenic organisms, and purification for domestic or industrial use is not necessary. Furthermore, groundwater supplies are not seriously affected by short droughts and are available in many areas that do not have dependable surface water supplies. However, aquifers and other groundwater supplies are at risk of chemical pollution from fracking, agricultural chemicals, leaking or unfit landfills and septic tanks, and other point and nonpoint sources of pollution. Such contamination can render groundwater unfit for use and is expensive and difficult to clean up.
It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1065 2021-06-29 00:03:40
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
1043) Public utility
Public utility, enterprise that provides certain classes of services to the public, including common carrier transportation (buses, airlines, railroads, motor freight carriers, pipelines, etc.); telephone and telegraph; power, heat, and light; and community facilities for water, sanitation, and similar services. In most countries such enterprises are state-owned and state-operated, but in the United States they are mainly privately owned and are operated under close governmental regulation.
The classic explanation for the need to regulate public utilities is that they are enterprises in which the technology of production, transmission, and distribution almost inevitably leads to complete or partial monopoly—that they are, in a phrase, natural monopolies. The monopolistic tendency arises from economies of scale in the particular industry, from the large capital costs typical of such enterprises, from the inelasticity of demand among consumers of the service, from considerations of the excess capacity necessary to meet demand peaks, and other considerations. It is often also the case that the existence of competing parallel systems—of local telephones or natural gas, for example—would be inordinately expensive, wasteful, and inconvenient. Given the tendency to monopoly and the potential therefore of monopolistic pricing practices, public regulation has for more than a century been applied to certain classes of business.
In practice, regulation aims to ensure that the utility serves all who apply for and are willing and able to pay for its services, that it operates in a safe and adequate manner, that it serves all customers on equal terms, and that its rates are just and reasonable. All states have regulatory commissions, and the federal government has several, including the Interstate Commerce Commission, the Civil Aeronautics Board, the Federal Power Commission, the Federal Communications Commission, and the Securities and Exchange Commission.

It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1066 2021-06-30 00:22:24
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
1044) Radio and radar astronomy
Radio and radar astronomy, study of celestial bodies by examination of the radio-frequency energy they emit or reflect. Radio waves penetrate much of the gas and dust in space, as well as the clouds of planetary atmospheres, and pass through Earth’s atmosphere with little distortion. Radio astronomers can therefore obtain a much clearer picture of stars and galaxies than is possible by means of optical observation. The construction of ever larger antenna systems and radio interferometers and improved radio receivers and data-processing methods have allowed radio astronomers to study fainter radio sources with increased resolution and image quality.
In 1932 the American physicist Karl Jansky first detected cosmic radio noise from the centre of the Milky Way Galaxy while investigating radio disturbances that interfered with transoceanic telephone service. (The radio source at the centre of the Galaxy is now known as Sagittarius A.) The American amateur radio operator Grote Reber later built the first radio telescope at his home in Wheaton, Ill., and found that the radio radiation came from all along the plane of the Milky Way and from the Sun. For the first time, astronomers could observe objects in a new region of the electromagnetic spectrum outside that of visible light.
During the 1940s and ’50s, Australian and British radio scientists were able to locate a number of discrete sources of celestial radio emission that they associated with old supernovae (Taurus A, identified with the Crab Nebula) and active galaxies (Virgo A and Centaurus A) that later became to be known as radio galaxies.
In 1951, American physicists Harold Ewen and E.M. Purcell detected 21-cm radiation emitted by cold clouds of interstellar hydrogen atoms. This emission was later used to define the spiral arms of the Milky Way Galaxy and to determine the rotation of the Galaxy.
In the 1950s, astronomers at Cambridge University published three catalogs of astronomical radio sources. The last of these, the Third Cambridge Catalogue (or 3C), published in 1959, contained some sources, most notably 3C 273, that were identified with faint stars. In 1963 American astronomer Maarten Schmidt observed 3C 273 with an optical telescope and found that it was not a star in the Milky Way Galaxy but a very distant object nearly two billion light-years from Earth. Objects like 3C 273 were called quasi-stellar radio sources, or quasars.
Beginning in the late 1950s, radio studies of the planets revealed the existence of a greenhouse effect on Venus, intense Van Allen radiation belts surrounding Jupiter, powerful radio storms in Jupiter’s atmosphere, and an internal heating source deep within the interiors of Jupiter and Saturn.
Radio telescopes are also used to study interstellar molecular gas clouds. The first molecule detected by radio telescopes was hydroxyl (OH) in 1963. Since then about 150 molecular species have been detected, only a few of which can be observed at optical wavelengths. These include carbon monoxide, ammonia, water, methyl and ethyl alcohol, formaldehyde, and hydrogen cyanide, as well as some heavy organic molecules such as the amino acid glycine.
In 1964, Bell Laboratories scientists Robert Wilson and Arno Penzias detected the faint cosmic microwave background (CMB) signal left over from the original big bang, thought to have occurred 13.8 billion years ago. Subsequent observations of this CMB in the 1990s and 2000s with the Cosmic Background Explorer and the Wilkinson Microwave Anisotropy Probe satellites have detected fine-scale deviations from the smooth background that correspond to the initial formation of structure in the early universe.
Radio observations of quasars led to the discovery of pulsars (or pulsating radio stars) by British astronomers Jocelyn Bell and Antony Hewish in Cambridge, Eng., in 1967. Pulsars are neutron stars that spin very rapidly, up to nearly 1,000 times per second. Their radio emission is concentrated along a narrow cone, producing a series of pulses corresponding to the rotation of the neutron star, much like the beacon from a rotating lighthouse lamp. In 1974, using the Arecibo Observatory, American astronomers Joseph Taylor and Russell Hulse observed a binary pulsar (two pulsars in orbit around each other) and found that their orbital period was decreasing because of gravitational radiation at exactly the rate predicted by Albert Einstein’s theory of general relativity.
Using powerful radar systems, it is possible to detect radio signals reflected from nearby astronomical bodies such as the Moon, the nearby planets, some asteroids and comets, and the larger moons of Jupiter. Precise measurements of the time delay between the transmitted and reflected signal and the spectrum of the returned signal are used to precisely measure the distance to solar system objects and to image their surface features with a resolution of a few metres. The first successful detection of radar signals from the Moon occurred in 1946. This was quickly followed by experiments in the United States and the Soviet Union using powerful radar systems built for military and commercial applications. Both radio and radar studies of the Moon revealed the sandlike nature of its surface even before the Apollo landings were made. Radar echoes from Venus have penetrated its dense cloud cover surrounding the surface and have uncovered valleys and enormous mountains on the planet’s surface. The first evidence for the correct rotation periods of Venus and of Mercury also came from radar studies.

It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1067 2021-07-01 00:25:08
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
1045) Mercury (chemical element)
Mercury (Hg), also called quicksilver, chemical element, liquid metal of Group 12 (IIb, or zinc group) of the periodic table.
Element Properties :
atomic number : 80
atomic weight : 200.59
melting point : −38.87 °C (−37.97 °F)
boiling point : 356.9 °C (674 °F)
specific gravity : 13.5 at 20 °C (68 °F)
valence : 1, 2
Properties, Uses, And Occurrence
Mercury was known in Egypt and also probably in the East as early as 1500 BCE. The name mercury originated in 6th-century alchemy, in which the symbol of the planet was used to represent the metal; the chemical symbol Hg derives from the Latin hydrargyrum, “liquid silver.” Although its toxicity was recognized at an early date, its main application was for medical purposes.
Mercury is the only elemental metal that is liquid at room temperature. (Cesium melts at about 28.5 °C [83 °F], gallium at about 30 °C [86 °F], and rubidium at about 39 °C [102 °F].) Mercury is silvery white, slowly tarnishes in moist air, and freezes into a soft solid like tin or lead at −38.87 °C (−37.97 °F). It boils at 356.9 °C (674 °F).
It alloys with copper, tin, and zinc to form amalgams, or liquid alloys. An amalgam with silver is used as a filling in dentistry. Mercury does not wet glass or cling to it, and this property, coupled with its rapid and uniform volume expansion throughout its liquid range, makes it useful in thermometers. Barometers and manometers utilize its high density and low vapour pressure. Gold and silver dissolve readily in mercury, and in the past this property was used in the extraction of these metals from their ores.
The good electrical conductivity of mercury makes it exceptionally useful in sealed electrical switches and relays. An electrical discharge through mercury vapour contained in a fused silica tube or bulb produces a bluish glow rich in ultraviolet light, a phenomenon exploited in ultraviolet, fluorescent, and high-pressure mercury-vapour lamps. Mercury’s high thermal neutron-capture cross section (360 barns) and good thermal conductivity make it applicable as a shield and coolant in nuclear reactors. Much mercury is utilized in the preparation of pharmaceuticals and agricultural and industrial fungicides.
The use of mercury in the manufacture of chlorine and caustic soda (sodium hydroxide) by electrolysis of brine depends upon the fact that mercury employed as the negative pole, or cathode, dissolves the sodium liberated to form a liquid amalgam. An interesting application, though not of great commercial significance, is the use of mercury vapour instead of steam in some electrical generating plants, the higher boiling point of mercury providing greater efficiency in the heat cycle.
Mercury occurs in Earth’s crust on the average of about 0.08 gram (0.003 ounce) per ton of rock. The principal ore is the red sulfide, cinnabar. Native mercury occurs in isolated drops and occasionally in larger fluid masses, usually with cinnabar, near volcanoes or hot springs. Over two-thirds of the world supply of mercury comes from China, with most of the remainder coming from Kyrgyzstan and Chile; it is often a by-product of gold mining. Cinnabar is mined in shaft or open-pit operations and refined by flotation. Most of the methods of extraction of mercury rely on the volatility of the metal and the fact that cinnabar is readily decomposed by air or by lime to yield the free metal. Because of the toxicity of mercury and the threat of rigid pollution control, attention is being directed toward safer methods of extracting mercury. These generally rely on the fact that cinnabar is readily soluble in solutions of sodium hypochlorite or sulfide, from which the mercury can be recovered by precipitation with zinc or aluminum or by electrolysis.
Extremely rare natural alloys of mercury have also been found: moschellandsbergite (with silver), potarite (with palladium), and gold amalgam. Mercury is extracted from cinnabar by roasting it in air, followed by condensation of the mercury vapour. Mercury is toxic. Poisoning may result from inhalation of the vapour, ingestion of soluble compounds, or absorption of mercury through the skin.
Natural mercury is a mixture of seven stable isotopes: 196Hg (0.15 percent), 198Hg (9.97 percent), 199Hg (16.87 percent), 200Hg (23.10 percent), 201Hg (13.18 percent), 202Hg (29.86 percent), and 204Hg (6.87 percent). As a wavelength standard and for other precise work, isotopically pure mercury consisting of only mercury-198 is prepared by neutron bombardment of natural gold, gold-197.
Principal Compounds
The compounds of mercury are either of +1 or +2 oxidation state. Mercury(II) or mercuric compounds predominate. Mercury does not combine with oxygen to produce mercury(II) oxide, HgO, at a useful rate until heated to the range of 300 to 350 °C (572 to 662 °F). At temperatures of about 400 °C (752 °F) and above, the reaction reverses with the compound decomposing into its elements. Antoine-Laurent Lavoisier and Joseph Priestley used this reaction in their study of oxygen.
There are relatively few mercury(I) or mercurous compounds. The mercury(I) ion, Hg22+, is diatomic and stable. Mercury(I) chloride, Hg2Cl2 (commonly known as calomel), is probably the most important univalent compound. It is used in antiseptic salves. Mercury(II) chloride, HgCl2 (also called bichloride of mercury or corrosive sublimate), is perhaps the commonest bivalent compound. Although extremely toxic, this odourless, colourless substance has a wide variety of applications. In agriculture it is used as a fungicide; in medicine it is sometimes employed as a topical antiseptic in concentrations of one part per 2,000 parts of water; and in the chemical industry it serves as a catalyst in the manufacture of vinyl chloride and as a starting material in the production of other mercury compounds. Mercury(II) oxide, HgO, provides elemental mercury for the preparation of various organic mercury compounds and certain inorganic mercury salts. This red or yellow crystalline solid is also used as an electrode (mixed with graphite) in zinc-mercuric oxide electric cells and in mercury batteries. Mercury(II) sulfide, HgS, is a black or red crystalline solid used chiefly as a pigment in paints, rubber, and plastics.

It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1068 2021-07-02 00:43:04
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
1046) Tungsten processing
Tungsten processing, preparation of the ore for use in various products.
Tungsten exhibits a body-centred cubic (bcc) crystal lattice. It has the highest melting point of all metals, 3,410° C (6,170° F), and it has high conductivity for electricity. Owing to this unique combination of properties, it is used extensively as filaments for incandescent lamps, as electric contacts, and as electron emitters for electronic devices. Tungsten also has found wide application as an alloying element for tool steels and wear-resistant alloys. Tungsten carbides are used for cutting tools and hard-facing materials owing to their hardness and resistance to wear. The metal is brittle at room temperature but ductile and strong at elevated temperatures. Its alloys are employed in rocket-engine nozzles and other aerospace applications.
History
Tungsten in one of its mineral forms was given its name (meaning “heavy stone”) by the Swedish mineralogist A.F. Cronstedt in 1755. In 1781 another Swede, Carl Wilhelm Scheele, analyzed the mineral and identified lime and an acid that he called tungstic acid; the mineral was later named scheelite. In 1783 the Spanish chemists Juan José and Fausto Elhuyar obtained metallic tungsten by the reduction of its oxide with carbon; it was named wolfram (hence its chemical symbol, W) for the mineral wolframite, from which it was extracted. In 1847, Robert Oxland patented in Britain his manufacturing process for sodium tungstate, tungstic acid, and the pure metal, and in 1857, he patented his process for producing tungsten steel. But it was not until 1908, when William David Coolidge obtained his British patent for producing ductile tungsten wire, that the filament industry began. Tungsten-containing high-speed tool steel came to public attention when the Bethlehem Steel Company exhibited its products at the Exposition Universelle of 1900 in Paris. In 1927 the Krupp Laboratory at Essen, Ger., discovered that a serviceable product could be produced when the normally brittle tungsten carbide was mixed with a cemented material.
Ores
Major minerals of tungsten are essentially of two categories. The first is wolframite [(Fe, Mn)WO4], which contains iron and manganese tungstates in all proportions between 20 and 80 percent of each. The second is scheelite (CaWO4), which fluoresces a bright bluish colour under ultraviolet light.
Tungsten deposits occur in association with metamorphic rocks and granitic igneous rocks. The most important mines are in the Nan Mountains in the Kiangsi, Hunan, and Kwangtung provinces of China, which possesses about 50 percent of the world’s reserves. In Russia, mines are located in the northern Caucasus and around Lake Baikal. There are also deposits in Kazakhstan. About 90 percent of South Korea’s tungsten is at Sang Dong. Canada’s Northwest Territories is home to the largest tungsten mine in the Western world, and a mine at Chojlla, Bol., is the largest producer in South America. Deposits in the United States are spread along the Rocky Mountains.
Mining And Concentrating
The Nan Mountains deposits are principally high-grade wolframite veins that are found outcropping in great numbers in many separate areas. These conditions are favourable for exploitation by small-scale operations. Open-pit methods have been used in Australia and Canada, while underground mining is generally necessary for other mines in the world.
Tungsten ores are beneficiated by crushing followed by gravity concentration. Flotation separation is used for scheelite that has been ground to a fine size to liberate the tungsten; this is further supplemented by leaching, roasting, and magnetic or high-tension separation when required.
Extraction And Refining
Ammonium paratungstate
Tungsten ores frequently occur in association with sulfides and math, which can be removed by roasting in air for two to four hours at 800° C (1,450° F). In order to produce ammonium paratungstate (APT), an intermediate compound in production of the pure metal, ores may be decomposed by acid leaching or by the autoclave-soda process. In the latter process, the ground ore is maintained for 11/2 to 4 hours in a solution of 10–18 percent sodium carbonate at temperatures of 190° to 230° C (375° to 445° F) and under a pressure of 14.1–24.6 kilograms per square centimetre (200–350 pounds per square inch). Prior to the removal of unreacted gangue by filtration, the acidity is adjusted to pH 9–9.5, and aluminum and manganese sulfates are added at 70°–80° C (160°–175° F) and stirred for one hour. This can eliminate phosphorus and math and reduce silica to a level of 0.03–0.06 percent. Molybdenum is removed by adding sodium sulfide at 80°–85° C (175°–185° F) at a pH of 10, holding for one hour, and then acidifying the solution to pH 2.5–3 and stirring for seven to nine hours to precipitate molybdenum sulfide. The remaining sodium tungstate solution can be further purified by a liquid ion-exchange process, using an organic extractant consisting of 7 percent alamine-336, 7 percent decanol, and 86 percent kerosene. During the countercurrent flow of the extractant through the solution, tungstate ions transfer from the aqueous phase to the organic phase. The tungsten is then stripped from the extractant into an ammonia solution containing ammonium tungstate. The resultant APT solution is sent to an evaporator for crystallization.
In the acid-leaching process, scheelite concentrate is decomposed by hydrochloric acid in the presence of sodium nitrate as an oxidizing agent. This charge is agitated by steam spraying and is maintained at 70° C (160° F) for 12 hours. The resultant slurry, containing tungsten in the form of a solid tungstic acid, is diluted and allowed to settle. The tungstic acid is then dissolved in aqueous ammonia at 60° C (140° F) for two hours under stirring. Calcium from the resulting solution is precipitated as calcium oxalate, while phosphorus and math may be removed by the addition of magnesium oxide, which forms insoluble phosphates and math of ammonium and magnesium. Iron, silica, and similar impurities that form colloidal hydroxides are removed by adding a small amount of activated carbon and digesting for one to two hours. The solution is clarified through pressure filters and evaporated to obtain APT crystals.
Tungsten powder
When APT is decomposed to tungsten oxides, it displays different colours according to its composition: the trioxide is yellow, the dioxide is brown, and the intermediate oxide is purple-blue. APT can be decomposed to yellow oxide when heated to above 250° C (480° F) in a furnace under a flow of air. In the industrial production of tungsten, however, APT is usually decomposed to the intermediate oxide in a rotary furnace under a stream of hydrogen, which partially decomposes the ammonia in the crystals into nitrogen and hydrogen while maintaining a reducing atmosphere. The rotary furnace is divided by partitions into three zones maintained, respectively, at 850°, 875°, and 900° C (1,550°, 1,600°, and 1,650° F). The furnace is tilted at a small angle and rotated to provide a continuous flow of powder through the central holes of the partitions.
The blue oxide is then reduced by hydrogen to metallic tungsten powder in stationary furnaces at temperatures ranging from 550° to 850° C (1,025° to 1,550° F). In this process the oxide is loaded into “boats” made of Inconel, a nickel-based alloy noted for its strength at high temperatures. These are stoked into tubes, usually arranged in two rows, and the tubes are heated in three separate zones along their lengths.
APT may also be reduced by carbon, although the powder is usually contaminated with tungsten carbide and some mineral elements contained in the carbon. When APT and carbon are mixed and reacted at 650°–850° C (1,200°–1,550° F), the product is a blue oxide. When heated in the range of 900°–1,050° C (1,650°–1,925° F), the brown oxide is formed. For complete reduction to metal, a temperature higher than 1,050° C is required. The purity of the metal is about 95 percent.
Consolidation
Tungsten powder is compacted into bars or billets with a mechanical or isostatic press prior to sintering. The “green,” or unfired, density of these compacts, obtained from powder particle sizes ranging from 1 to 10 micrometres, is usually 65 to 75 percent of the theoretical. After being presintered at 1,000°–1,200° C (1,800°–2,200° F), tungsten bars of small diameter are sintered in a hydrogen atmosphere, with heat being provided by the direct-resistance method—that is, by an electric current passed through the bar. A spring attachment to the water-cooled clips holding each bar is necessary so that one end is free to move as the bar shrinks during sintering. The current is gradually increased to raise the temperature from room temperature to 2,700°–3,100° C (4,900°–5,600° F). After holding at the final temperature for 30 to 60 minutes, the density reaches 88.5 to 96 percent of the theoretical.
An indirect sintering process is used for large tungsten billets. The heating elements of the furnace are constructed of molybdenum strips and supported by molybdenum or tungsten frames, and they are surrounded by molybdenum heat shields. A slow heating in the early stage of sintering is essential for deoxidizing the material and releasing gases at a controlled rate. At higher temperatures—i.e., from 800° C up to the final sintering temperature of 2,400° C (4,350° F)—the heating rate also should be controlled, since too fast a temperature buildup within the billet would cause thermal stresses and would result in the cracking of the material. A final sintering for 10 hours is required for densification.
The Metal And Its Alloys
Tungsten filaments doped with approximately 0.05 percent each of alumina, silica, and potassium oxide exhibit nonsagging behaviour and are used in incandescent lamps. Adding 1 to 2 percent thoria or zirconia increases the electron emission and high-temperature strength of tungsten wire, making it useful for electronic applications and electrodes for tungsten–inert-gas arc welding.
Tungsten infiltrated by silver and copper has excellent arc resistance, high resistance to welding, and high conductivity and current capacity. Consequently, it is widely used for electrical contacts, semiconductor supports, and rocket nozzles.
Tungsten is an important addition to tool steels, superalloys, and refractory alloys. Cobalt-chromium-tungsten alloys, produced under the trade name Stellites, are used for the hard-facing of wear-resistant valves, bearings, propeller shafts, cutting tools, and high-temperature tools.
Chemical Compounds
Tungsten carbide (WC)
Tungsten carbides are divided into two categories. The first is the cemented tungsten carbides, also called hard metals, which are essentially WC produced from sintering a mixture of carbon black and hydrogen-reduced tungsten powder at 1,500° C (2,700° F). These are cemented using a cobalt or nickel binder, with or without other refractory carbides. The major uses of cemented carbides are for cutting and drilling tools, forming and drawing dies, and tire studs.
The second group is called fused or cast carbide, consisting of W2C and a eutectic mixture of WC and W2C. Harder but more brittle than the cemented carbide, it is used in wear-resisting applications such as anvils, guide sleeves in machines, and teeth and jaws for excavators.
Other compounds
Tungsten bronze, composed of tungstates of the alkali and alkaline-earth metals, is employed as a substitute for bronze in ornamental paints. Sodium tungstate is also used to produce phosphotungstic acid-type organic dyes and pigments, which are brilliant, light-resistant, and insoluble in water and linseed oil. Calcium and magnesium tungstates are used as phosphors in fluorescent light and television tubes. Ammonium tungstate and other compounds are used as catalysts in the petroleum industry for hydrotreating, hydrocracking, and polymerization.

It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1069 2021-07-03 00:04:41
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
1047) Vanadium processing
Vanadium processing, preparation of the metal for use in various products.
Vanadium (V) is a grayish silver metal whose crystal structure is a body-centred cubic (bcc) lattice, with a melting point of 1,926° C (3,499° F). The metal is used principally as an alloying addition to high-strength low-alloy (HSLA) steels and, to a lesser extent, in tool steels and iron and steel castings. It is also an important strengthener for titanium alloys. Vanadium alloys are promising candidates for applications in nuclear reactors. The metal is recognized as an industrial hazard, however, as breathing of particulate material with a high vanadium content has been observed to cause an intense, dry cough accompanied by irritation of the nose, eyes, and throat.
History
The discovery of vanadium was first claimed in 1801 by a Spanish mineralogist, Andrés Manuel del Río, who gave it the name erythronium, after the red colour of one of its chemical compounds (Greek erythros, “red”). In 1830 a Swedish chemist, Nils Gabriel Sefström, rediscovered the element and named it vanadium, after Vanadis, the Scandinavian goddess of beauty, because of the beautiful colours of its compounds in solution. The English chemist Henry Enfield Roscoe first isolated the metal by hydrogen reduction of vanadium dichloride in 1867, and the American chemists John Wesley Marden and Malcolm N. Rich obtained vanadium of 99.7 percent purity by a calcium reduction process in 1925.
Since the early 1900s, vanadium has been used as an alloying element for steels and iron. In 1905 Antenor Riza Patron discovered a large asphaltite deposit containing rich vanadium ores in Mina Ragra, Peru. Two years later, the American Vanadium Company produced ferrovanadium on a commercial scale for the first time. After titanium became an aerospace construction material in the 1950s, vanadium saw wide use in titanium alloys.
Ores
The important vanadium minerals are patronite (VS4), carnotite [K2(UO2)2(VO4)2], and vanadinite, [Pb5(VO4)3Cl]. Ore deposits mined solely for vanadium are rare because much of the vanadium in igneous rocks occurs in the relatively insoluble trivalent state, substituting for ferric iron in ferromagnesium silicates, magnetite (an iron ore), ilmenite (a titanium ore), and chromite.
The world’s largest mines of vanadium are from titaniferous magnetite reserves in such regions as the Bushveld of South Africa, the Kachkanar Massif of the Ural Mountains, and China’s Szechwan province. Carnotite ores in the sandstones of the Colorado Plateau have been mined for vanadium and uranium. Other sources of vanadium include ash from the combustion of fossil fuel, slag from phosphate ore, the aluminum ore bauxite, and spent catalysts.
Mining
Because vanadium is essentially the by-product of ores that are mined for other minerals, they are mined by methods peculiar to those ores.
Extraction And Refining
Vanadium pentoxide
Titaniferous magnetite ore is partially reduced with coal in rotary kilns and then melted in a furnace. This produces a slag containing most of the titanium and a pig iron containing most of the vanadium. After removing the slag, the molten pig iron is blown with oxygen to form a new slag containing 12–24 percent vanadium pentoxide (V2O5), which is used in the further processing of the metal.
Vanadium is extracted from carnotite as a coproduct with uranium by leaching the ore concentrate for 24 hours with hot sulfuric acid and an oxidant such as sodium chlorate. After removal of solids, the leachate is fed into a solvent extraction circuit where the uranium is extracted in an organic solvent consisting of 2.5-percent-amine–2.5-percent-isodecanol–95-percent-kerosene. Vanadium remains in the raffinate, which is fed into a second solvent extraction circuit. There vanadium in turn is extracted in the organic phase, stripped with a 10 percent soda ash solution, and precipitated with ammonium sulfate. The ammonium metavanadate precipitate is filtered, dried, and calcined to V2O5.
Most other vanadium-bearing ores or slags are crushed, ground, screened, and mixed with a sodium salt such as sodium chloride or sodium carbonate. This charge is then roasted at about 850° C (1,550° F) to convert the oxides to sodium metavanadate, which can be leached in hot water. With the acidulation of the leachate with sulfuric acid, the vanadium is precipitated as sodium hexavanadate. This compound, known as red cake, can be fused at 700° C (1,300° F) to yield technical-grade vanadium pentoxide (at least 86 percent V2O5, or it can be further purified by dissolving it in an aqueous solution of sodium carbonate. In the latter case, the iron, aluminum, and silicon impurities in the red cake precipitate from solution upon adjustment of the acidity. The vanadium is precipitated as ammonium metavanadate by adding ammonium chloride. After filtration, the precipitate is calcined to produce V2O5 of a purity greater than 99.8 percent.
Ferrovanadium
The production of ferrovanadium, containing 35–80 percent vanadium, is carried out in an electric-arc furnace. Scrap iron is first melted, and a mixture of V2O5, aluminum, and a flux such as calcium fluoride or calcium oxide is added. In the ensuing reaction, the aluminum metal is converted to alumina, forming a slag, and the V2O5 is reduced to vanadium metal, which is dissolved in the molten iron. Since this oxidation-reduction reaction is exothermic, the heat supply need only develop the kindling temperature of 950° C (1,750° F). After kindling, the electrodes are withdrawn until the reaction is completed; they are then reinserted into the molten slag and the furnace reheated to improve settling.
The aluminothermic process can also be carried out in a refractory-lined steel pot or water-cooled copper crucible. A charge of V2O5, iron oxide, and aluminum is ignited with a barium-peroxide fuse or a magnesium ribbon.
Vanadium metal
In the production of pure metal, V2O5 is reduced metallothermically by calcium or aluminum. In the calcium reduction, the exothermic reaction is carried out in a sealed vessel using calcium chloride as a flux. The vanadium metal is recovered in the form of droplets or beads. (A massive regulus can be obtained by using iodine as both a flux and a thermal booster.) The calcium process requires a rather large amount of reductant and gives low metal yields—in the range of 75–80 percent. In the aluminothermic process, V205, mixed with aluminum powder, is heated in an electric furnace or ignited in a refractory-lined vessel using barium peroxide as the booster. The vanadium regulus thus obtained may be further purified by electron-beam melting.
To prepare aluminum-vanadium master alloys for the titanium industry, the aluminothermic method is also used. In this case, an amount of aluminum greater than that required for reduction is added to the charge.
The Metal And Its Alloys
In its pure form, vanadium is soft and ductile. It can be fabricated into mill forms, but it oxidizes readily at temperatures above 663° C (1,225° F) and is liable to pick up interstitial impurities. Because the metal has good corrosion resistance to liquid metal, a low absorption of neutrons, and a short half-life in its radioactive isotopic forms, vanadium-based alloys have potential as structural materials for fusion and liquid-metal fast-breeder fission reactors.
Iron and steel
The addition of small amounts of vanadium (less than 0.2 percent) to structural steels improves their toughness, ductility, and strength owing to the grain-refining effect of vanadium carbide precipitates. These HSLA steels are used in automotive components, such as hoods and door panels, and in oil and gas pipelines.
Almost all tool steels contain vanadium in amounts ranging from 0.10 to 5 percent. It is required to ensure the retention of hardness and cutting ability at high temperatures.
In some cast irons, the addition of a small amount of vanadium controls the size and distribution of graphite flakes, thereby improving strength and wear resistance. Steel castings with vanadium additions also exhibit pronounced shock and wear resistance, which makes them useful in heavy-duty equipment and machinery.
Titanium
Vanadium improves the strength of titanium alloys and promotes their thermal stability. Several important commercial titanium alloys contain between 2.5 and 15 percent vanadium. They are used in the undercarriages, wings, and engines of jet aircraft.
Chemical Compounds
Catalysts
Vanadium is used in the contact process for the manufacture of sulfuric acid. In this process, sulfur dioxide is oxidized to a trioxide by exposure to air in the presence of granular V2O5 or sodium metavanadate. Vanadium oxytrichloride and vanadium tetrachloride are catalysts in the production of special types of synthetic rubber. Ammonium metavanadate is employed as a catalyst for the synthesis of organic intermediates of nylon, polyester resins, and other synthetics, and it has also been used as a catalyst in the dyeing of leather and fur.
Pigments
In dye manufacturing, vanadium compounds are used in the production of aniline black. They are also employed as mordants in the dyeing and printing of cotton and for fixing aniline black on silk. Some modern quick-drying inks depend on the addition of ammonium metavanadate for their performance. Vanadium compounds are used in the ceramics industry for glazes and enamels.

It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1070 2021-07-04 01:07:37
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
1048) Sodium
Sodium (Na), chemical element of the alkali metal group (Group 1 [Ia]) of the periodic table. Sodium is a very soft silvery-white metal. Sodium is the most common alkali metal and the sixth most abundant element on Earth, comprising 2.8 percent of Earth’s crust. It occurs abundantly in nature in compounds, especially common salt—sodium chloride (NaCl)—which forms the mineral halite and constitutes about 80 percent of the dissolved constituents of seawater.
Element Properties
atomic number : 11
atomic weight : 22.9898
melting point : 97.81 °C (208 °F)
boiling point : 882.9 °C (1,621 °F)
specific gravity : 0.971 (20 °C)
oxidation states : +1, −1 (rare)
Properties And Production
Because sodium is extremely reactive, it never occurs in the free state in Earth’s crust. In 1807 Sir Humphry Davy became the first to prepare sodium in its elemental form, applying electrolysis to fused sodium hydroxide (NaOH). Sodium is an important constituent of a number of silicate materials, such as feldspars and micas. There are huge deposits of rock salt in various parts of the world, and sodium nitrate deposits exist in Chile and Peru. The sodium content of the sea is approximately 1.05 percent, corresponding to a concentration of approximately 3 percent of sodium halides. Sodium has been identified in both the atomic and ionic forms in the spectra of stars, including the Sun, and the interstellar medium. Analysis of meteorites indicates that the silicate material present has an average content of approximately 4.6 atoms of sodium for every 100 atoms of silicon.
Lighter than water, sodium can be cut with a knife at room temperature but is brittle at low temperatures. It conducts heat and electricity easily and exhibits the photoelectric effect (emission of electrons when exposed to light) to a marked degree.
Sodium is by far the most commercially important alkali metal. Most processes for the production of sodium involve the electrolysis of molten sodium chloride. Inexpensive and available in tank-car quantities, the element is used to produce gasoline additives, polymers such as nylon and synthetic rubber, pharmaceuticals, and a number of metals such as tantalum, titanium, and silicon. It is also widely used as a heat exchanger and in sodium-vapour lamps. The yellow colour of the sodium-vapour lamp and the sodium flame (the basis of an analytical test for sodium) is identified with two prominent lines in the yellow portion of the light spectrum.
Significant Uses
Two of the earliest uses of metallic sodium were in the manufacture of sodium cyanide and sodium peroxide. Significant quantities were used in the manufacture of tetraethyl lead as a gasoline additive, a market that disappeared with the advent of unleaded gasoline. Substantial amounts of sodium are used in the manufacture of sodium alkyl sulfates as the principal ingredient in synthetic detergents.
Sodium also is used as a starting material in the manufacture of sodium hydride (NaH) and sodium borohydride (NaBH4). In addition, sodium is employed in the production of dyes and dye intermediates, in the synthesis of perfumes, and in a wide variety of organic reductions. It is used in the purification of hydrocarbons and in the polymerization of unsaturated hydrocarbons. In many organic applications, sodium is used in the form of dispersions in hydrocarbon liquid media.
Molten sodium is an excellent heat-transfer fluid, and, because of this property, it has found use as coolant in liquid-metal fast breeder reactors. Sodium is used extensively in metallurgy as a deoxidant and as a reducing agent for the preparation of calcium, zirconium, titanium, and other transition metals. Commercial production of titanium involves reduction of titanium tetrachloride (TiCl4) with sodium. The products are metallic Ti and NaCl.
Principal Compounds
Sodium is highly reactive, forming a wide variety of compounds with nearly all inorganic and organic anions (negatively charged ions). It normally has an oxidation state of +1, and its single valence electron is lost with great ease, yielding the colourless sodium cation (Na+). Compounds that contain the sodium anion, Na−, have also been synthesized. The principal commercial sodium compounds are the chloride, carbonate, and sulfate.
The most important and familiar sodium compound is sodium chloride, or common salt, NaCl. Most other sodium compounds are prepared either directly or indirectly from sodium chloride, which occurs in seawater, in natural brines, and as rock salt. Large quantities of sodium chloride are employed in the production of other heavy (industrial) chemicals as well as being used directly for ice and snow removal, for water conditioning, and in food.
Other major commercial applications of sodium chloride include its use in the manufacture of chlorine and sodium hydroxide by electrolytic decomposition and in the production of sodium carbonate (Na2CO3) by the Solvay process. The electrolysis of aqueous sodium chloride produces sodium hypochlorite, NaOCl, a compound of sodium, oxygen, and chlorine used in large quantities in household chlorine bleach. Sodium hypochlorite is also utilized as an industrial bleach for paper pulp and textiles, for chlorination of water, and in certain medicinal preparations as an antiseptic and a fungicide. It is an unstable compound known only in aqueous solution.
The carbonates contain the carbonate ion (CO32–). Sodium bicarbonate, also called sodium hydrogen carbonate, or bicarbonate of soda, NaHCO3, is a source of carbon dioxide and so is used as an ingredient in baking powders, in effervescent salts and beverages, and as the main constituent of dry-chemical fire extinguishers. Its slight alkalinity makes it useful in treating gastric or urinary hyperacidity and acidosis. It is also employed in certain industrial processes, as in tanning and the preparation of wool. Sodium carbonate, or soda ash, Na2CO3, is widely distributed in nature, occurring as constituents of mineral waters and as the solid minerals natron, trona, and thermonatrite. Large quantities of this alkaline salt are used in making glass, detergents, and cleansers. Sodium carbonate is treated with carbon dioxide to produce sodium bicarbonate. The monohydrate form of sodium carbonate, Na2CO3•H2O, is employed extensively in photography as a constituent in developers.
Sodium sulfate, Na2SO4, is a white crystalline solid or powder employed in the manufacture of kraft paper, paperboard, glass, and detergents and as a raw material for the production of various chemicals. It is obtained either from deposits of the sodium sulfate minerals mirabilite and thenardite or synthetically by the treatment of sodium chloride with sulfuric acid. The crystallized product is a hydrate, Na2SO4•10H2O, commonly known as Glauber’s salt. Sodium thiosulfate (sodium hyposulfite), Na2S2O3, is used by photographers to fix developed negatives and prints; it acts by dissolving the part of the silver salts coated onto film which remain unchanged by exposure to light.
Sodium hydroxide (NaOH) is a corrosive white crystalline solid that readily absorbs moisture until it dissolves. Commonly called caustic soda, or lye, sodium hydroxide is the most widely used industrial alkali. It is highly corrosive to animal and vegetable tissue. The alkaline solutions it forms when dissolved in water neutralize acids in various commercial processes: in petroleum refining, it removes sulfuric and organic acids; in soapmaking, it reacts with fatty acids. Solutions of NaOH are used in the treatment of cellulose and in the manufacture of many chemicals.
Sodium nitrate, or soda nitre, NaNO3, is commonly called Chile saltpetre, after its mineral deposits in northern Chile, the principal source. Sodium nitrate is used as a nitrogenous fertilizer and as a component of dynamite.
Chemical Properties
Generally, elemental sodium is more reactive than lithium, and it reacts with water to form a strong base, sodium hydroxide (NaOH). Its chemistry is well explored.
Reaction with air, water, and hydrogen
Sodium is ordinarily quite reactive with air, and the reactivity is a function of the relative humidity, or water-vapour content of the air. The corrosion of solid sodium by oxygen also is accelerated by the presence of small amounts of impurities in the sodium. In ordinary air, sodium metal reacts to form a sodium hydroxide film, which can rapidly absorb carbon dioxide from the air, forming sodium bicarbonate. Sodium does not react with nitrogen, so sodium is usually kept immersed in a nitrogen atmosphere (or in inert liquids such as kerosene or naphtha). It is significantly more reactive in air as a liquid than as a solid, and the liquid can ignite at about 125 °C (257 °F). In a comparatively dry atmosphere, sodium burns quietly, giving off a dense white caustic smoke, which can cause choking and coughing. The temperature of burning sodium increases rapidly to more than 800 °C (1,500 °F), and under these conditions the fire is extremely difficult to extinguish. Special dry-powder fire extinguishers are required, since sodium reacts with carbon dioxide, a common propellant in regular fire extinguishers.
Sodium monoxide (Na2O) is ordinarily formed upon oxidation of sodium in dry air. The superoxide (NaO2) can be prepared by heating metallic sodium to 300 °C (570 °F) in an autoclave (a heated pressure vessel) containing oxygen at high pressure. Another route to the superoxide is oxidation of sodium peroxide, Na2O2, treated to have a large surface area.
Sodium that is heavily contaminated with the monoxide may be readily purified by filtration, since the solubility of the oxide in molten sodium is low. This low solubility is utilized to a considerable extent in continuous purification processes of the sodium in large liquid-metal reactor systems. A second technique for removing the oxide, called cold trapping, involves running the molten sodium through a cooled packed bed of material, upon which the oxide can precipitate. Filtration and cold trapping also are effective in removal of gross quantities of carbonate, hydroxide, and hydride.
The reaction with water of liquid sodium having a high surface area can be explosive. The sodium-water reaction is highly exothermic (that is, heat is given off).
Tests have indicated, however, that sodium and water cannot be mixed fast enough to produce the shock waves characteristic of high explosives. The explosive hazards of the reaction are associated primarily with the hydrogen gas that is formed.
Pure sodium begins to absorb hydrogen appreciably at about 100 °C (212 °F); the rate of absorption increases with temperature. Pure sodium hydride can be formed at temperatures above 350 °C (660 °F) by exposing sodium to hydrogen gas at a high flow rate. At higher temperatures the dissociation of sodium hydride to produce hydrogen and molten sodium is considerably greater than that of lithium hydride but slightly less than that of potassium hydride.
Reaction with nonmetals
Generally, alkali metals react with halogen gases, the degree of reactivity decreasing with increasing atomic weight of the halogen. Sodium is no exception to this statement. Under certain conditions of reaction, sodium and halogen vapours react to produce light (chemiluminescence). Halogen acids, such as hydrochloric acid, react vigorously with sodium, yielding the sodium halides. The reactions are highly exothermic, with heats of reaction (energy given off) of −71.8 and −76.2 kcal, respectively, for the reactions with hydrofluoric and hydrochloric acids. Sodium is attacked by other strong mineral acids to form the corresponding salts. It reacts with the fumes of nitric acid at 15 °C (59 °F) to form sodium nitrate and with acetic and sulfuric acids to form sodium acetate and sodium sulfate. With molten sulfur it reacts violently to produce polysulfides; under more controlled conditions it reacts with organic solutions of sulfur. Liquid selenium and tellurium both react vigorously with solid sodium to form selenides and tellurides.
Sodium shows relatively little reactivity with carbon, although lamellar (layerlike) materials can be prepared in which sodium is present between graphite layers. At 625 °C (1,157 °F) carbon monoxide reacts with sodium to form sodium carbide and sodium carbonate.
With the exception of the oxides of the Group 4 (IVb) metals (titanium, zirconium, and hafnium), the oxides of the transition metals are all reduced to the respective metals with elemental sodium. Sodium also reacts with a large number of metallic halides, displacing the metal from the salt and forming a sodium halide in the process. This reaction is used in the preparation of several of the transition metals themselves, including titanium and tantalum.
Sodium and all the other alkali metals dissolve in liquid ammonia to give intense blue solutions, and at ordinary temperatures a slow reaction between sodium and ammonia occurs to form sodamide, NaNH2, and hydrogen, similar to the reaction of sodium with water to give NaOH and hydrogen.
The reaction of alkali metal-ammonia solutions to form the amide and hydrogen can be catalyzed by the addition of many metals and metal oxides.
Liquid ammonia is often used as a solvent for sodium, allowing a number of reactions to occur at ordinary temperatures that would otherwise need heat. Sodium superoxide (NaO2), for example, can be formed by passing oxygen through ammonia solutions of sodium at −77 °C (−107 °F). Ammonia also serves as a solvent for reactions of sodium with math, tellurium, antimony, bismuth, and a number of other low-melting metals. Sodium-ammonia solutions are used to blacken polytetrafluoroethylene (Teflon) to prepare its surface for cementing to other materials. The high reducing power of sodium-ammonia solutions makes them useful in a number of organic reactions known as Birch reductions.
Organic reactions
The organic reactions of sodium have been studied to a greater extent than those of any of the other alkali metals. Sodium reacts with anhydrous alcohols to form the respective alcoholates (or alkoxides).
The reaction is most vigorous with methanol and decreases with increasing molecular weight of the alcohol. Sodium methoxide is produced on an industrial scale by reaction of sodium with excess methanol. Organic acids react with sodium to form sodium salts.
The large negative free energy of formation of sodium halides permits the dehalogenation of a number of organic halides, the formation of the sodium halide being energetically favoured. The so-called Wurtz reaction—based on this principle—is used in organic synthesis to a considerable extent:
2RCl + 2Na → R―R + 2NaCl.
By this reaction, octane can be made from bromobutane and sodium. Organosodium compounds include a number in which the sodium atom is bonded directly to a carbon atom; an example is methylsodium, Na―CH3.
Sodium reacts violently with a number of halogenated hydrocarbons. For example, a violent explosion occurs when a mixture of carbon tetrachloride and sodium is subjected to shock. Even when the sodium is diluted to a considerable extent—as in sodium amalgam—a brisk reaction with carbon tetrachloride occurs.
Reaction with metals
Sodium is completely miscible with the alkali metals below it in the periodic table (potassium, rubidium, and cesium). A eutectic (that is, an alloy that melts lower than its components) melting at −10 °C (14 °F) is formed in the sodium-potassium system and is known commercially as NaK. Its composition is approximately 78 percent potassium, and it is used as a heat-transfer fluid and as an organic reactant. The eutectics formed in the sodium-rubidium and sodium-cesium binary systems melt, respectively, at −4.5 and −30 °C (24 and −22 °F). Sodium is the minor component with potassium and cesium of the ternary alloy NaKCs, melting at −78 °C (−108 °F). This fluid is the lowest-melting liquid alloy yet isolated.
Sodium also forms alloys with the alkaline-earth metals. Beryllium is soluble in sodium only to the extent of a few atomic percent at approximately 800 °C (1,500 °F). Liquid sodium and magnesium are only partially miscible. The degree of solubility in sodium of the alkaline-earth metals increases with increasing atomic weight, with the result that the solubility of calcium is 10 percent by weight at 700 °C (1,300 °F). In the sodium-strontium system, there is a considerable degree of miscibility. Sodium forms a number of compounds with barium, and several eutectics exist in the system.
The precious metals, such as silver, gold, platinum, palladium, and iridium, and the white metals, such as lead, tin, bismuth, and antimony, alloy to an appreciable extent with liquid sodium. Cadmium and mercury also react with sodium, and a number of compounds exist in both binary systems. Seven sodium-mercury compounds, or amalgams, exist, with Hg2Na having the highest melting point (354 °C, or 669 °F). Sodium amalgams are used chiefly for carrying out reactions in situations in which pure elemental sodium would be violently reactive and difficult to control. The solubility of transition metals in alkali metals is generally very low, often in the 1–10-parts per million range even at temperatures in excess of 500 °C (930 °F).
Nuclear Properties
Natural sodium is the stable isotope of mass 23. Of the radioactive artificial isotopes, sodium-22 (2.6-year half-life, the longest half-life of a sodium isotope) is used as a radioactive tracer for natural sodium. Sodium-24 (15-hour half-life) is limited in use by its short life and is produced by irradiation in a nuclear reactor. Because of this reaction, a sodium-cooled reactor must have a second heat-transfer loop so that radioactive sodium does not come in contact with the environment. Other isotopes have half-lives of a minute or less.
Biological Properties
Sodium salts, particularly sodium chloride, are found almost everywhere in biological material. Sodium is an essential element for life, as is potassium, and the two elements maintain a definite balance within the cell structure. Electrolyte balance between the inside of the cell and the outside is maintained by “active transport” of potassium ions into the cell and sodium ions out of the cell. Most of the biological effects of sodium salts are the result of the cation (Na+), with the negative counter-ion apparently not playing a dominant role.
The presence of salinity in soils is often detrimental to plant growth. Sodium ions replace calcium and other ions in clay complexes, transforming the clay to a sticky mass; water percolation is then drastically reduced, and the basicity of the soil rises markedly.
The tolerance of fish to changes in salinity is often quite remarkable. Many marine bacteria and diatoms are able to tolerate salt concentrations as great as 25 percent. The minimum sodium requirement for mammals appears to be 0.05 percent of the diet, corresponding in a normal adult to a requirement of 1–2 grams (0.04–0.07 ounce) of salt per day, which results in an average sodium content of body tissues of 0.24 percent. There is a wide variation of sodium content in the different tissues, with whole blood containing approximately 0.62 percent sodium chloride, whereas skin has a sodium content of less than 0.1 percent. There is a relationship between salt content and water balance of the body; a low salt intake causes loss of water. Considerable quantities of sodium are lost through the skin by perspiration, and considerable quantities can be excreted in the urine.

It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1071 2021-07-05 00:19:25
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
1049) Lead
Lead (Pb), a soft, silvery white or grayish metal in Group 14 (IVa) of the periodic table. Lead is very malleable, ductile, and dense and is a poor conductor of electricity. Known in antiquity and believed by the alchemists to be the oldest of metals, lead is highly durable and resistant to corrosion, as is indicated by the continuing use of lead water pipes installed by the ancient Romans. The symbol Pb for lead is an abbreviation of the Latin word for lead, plumbum.
Element Properties
atomic number : 82
atomic weight : 207.19
melting point : 327.5 °C (621.5 °F)
boiling point : 1,744 °C (3,171.2 °F)
density : 11.29 gram/cm³ at 20 °C (68 °F)
oxidation states : +2, +4
Occurrence And Distribution
Lead is mentioned often in early biblical accounts. The Babylonians used the metal as plates on which to record inscriptions. The Romans used it for tablets, water pipes, coins, and even cooking utensils; indeed, as a result of the last use, lead poisoning was recognized in the time of Augustus Caesar. The compound known as white lead was apparently prepared as a decorative pigment at least as early as 200 BCE. Modern developments date to the exploitation in the late 1700s of deposits in the Missouri-Kansas-Oklahoma area in the United States.
On a weight basis, lead has nearly the same abundance in Earth’s crust as tin. Cosmically, there is 0.47 lead atom per 106 silicon atoms. The cosmic abundance is comparable to those of cesium, praseodymium, hafnium, and tungsten, each of which is regarded as a reasonably scarce element.
Although lead is not abundant, natural concentration processes have resulted in substantial deposits of commercial significance, particularly in the United States but also in Canada, Australia, Spain, Germany, Africa, and South America. Significant deposits are found in the United States in the western states and the Mississippi valley. Rarely found free in nature, lead is present in several minerals, but all are of minor significance except the sulfide, PbS (galena, or lead glance), which is the major source of lead production throughout the world. Lead is also found in anglesite (PbSO4) and cerussite (PbCO3). By the early 21st century, China, Australia, the United States, Peru, Mexico, and India were the world’s top producers of lead in concentrate.
Lead may be extracted by roasting the ore and then smelting it in a blast furnace or by direct smelting without roasting. Additional refining removes impurities present in the lead bullion produced by either process. Almost half of all refined lead is recovered from recycled scrap.
Uses Of The Metal
Only a single crystalline modification, with a close-packed metallic lattice, is known. Properties that are responsible for the many uses of elemental lead include its ductility, ease of welding, low melting point, high density, and ability to absorb gamma radiation and X-radiation. Molten lead is an excellent solvent and collector for elemental silver and gold. The structural applications of lead are limited by its low tensile and fatigue strengths and its tendency to flow even when only lightly loaded.
When freshly cut, lead oxidizes quickly, forming a dull gray coating, formerly thought to be lead suboxide, Pb2O, but now recognized as a mixture of lead and lead monoxide, PbO, which protects the metal from further corrosion. Similarly, although lead is soluble in dilute nitric acid, it is only superficially attacked by hydrochloric or sulfuric acids because the insoluble chloride (PbCl2) or sulfate (PbSO4) coatings that are formed prevent continued reaction. Because of this general chemical resistance, considerable amounts of lead are used in roofing, as coverings for electric cables placed in the ground or underwater, and as linings for water pipes and conduits and structures for the transportation and processing of corrosive substances.
Elemental lead can also be oxidized to the Pb2+ ion by hydrogen ions, but the insolubility of most salts of Pb2+ makes lead resistant to attack by many acids. Oxidation under alkaline conditions is easier to effect and is favoured by the formation of the soluble species of lead in the +2 oxidation state. Lead oxide (PbO2, with lead as the Pb4+ ion) is among the stronger oxidizing agents in acidic solution, but it is comparatively weak in alkaline solution. The ease of oxidation of lead is enhanced by complex formation. The electrodeposition of lead is best effected from aqueous solutions containing lead hexafluorosilicate and hexafluorosilicic acid.
Lead has many other applications, the largest of which is in the manufacture of storage batteries. It is used in ammunition (shot and bullets) and as a constituent of solder, type metal, bearing alloys, fusible alloys, and pewter. In heavy and industrial machinery, sheets and other parts made from lead compounds may be used to dampen noise and vibration. Because lead effectively absorbs electromagnetic radiation of short wavelengths, it is used as a protective shielding around nuclear reactors, particle accelerators, X-ray equipment, and containers used for transporting and storing radioactive materials. Together with the compound lead oxide (PbO2) and with lead-antimony or lead-calcium alloys, it is employed in common storage batteries.
Properties Of The Element
Lead and its compounds are toxic and are retained by the body, accumulating over a long period of time—a phenomenon known as cumulative poisoning—until a lethal quantity is reached. The toxicity of lead compounds increases as their solubility increases. In children the accumulation of lead may result in cognitive deficits; in adults it may produce progressive renal disease. Symptoms of lead poisoning include abdominal pain and diarrhea followed by constipation, nausea, vomiting, dizziness, headache, and general weakness. Elimination of contact with a lead source is normally sufficient to effect a cure. The elimination of lead from insecticides and paint pigments and the use of respirators and other protective devices in areas of exposure have reduced lead poisoning materially. The recognition that the use of tetraethyl lead, Pb(C2H5)4, as an antiknock additive in gasoline was polluting the air and water led to the compound’s elimination as a gasoline constituent in the 1980s.
Nuclear Properties
Lead is formed both by neutron-absorption processes and the decay of radionuclides of heavier elements. Lead has four stable isotopes; their relative abundances are lead-204, 1.48 percent; lead-206, 23.6 percent; lead-207, 22.6 percent; and lead-208, 52.3 percent. Three stable lead nuclides are the end products of radioactive decay in the three natural decay series: uranium (decays to lead-206), thorium (decays to lead-208), and actinium (decays to lead-207). More than 30 radioactive isotopes have been reported. Of the radioactive isotopes of lead, the following appear as members of the three natural decay series: (1) thorium series: lead-212; (2) uranium series: lead-214 and lead-210; (3) actinium series: lead-211. The atomic weight of natural lead varies from source to source, depending on its origin by heavier element decay.
Compounds
Lead shows oxidation states of +2 and +4 in its compounds. Among the many important lead compounds are the oxides: lead monoxide, PbO, in which lead is in the +2 state; lead dioxide, PbO2, in which lead is in the +4 state; and trilead tetroxide, Pb3O4. Lead monoxide exists in two modifications, litharge and massicot. Litharge, or alpha lead monoxide, is a red or reddish yellow solid, has a tetragonal crystal structure, and is the stable form at temperatures below 488 °C (910 °F). Massicot, or beta lead monoxide, is a yellow solid and has an orthorhombic crystal structure; it is the stable form above 488 °C. Both forms are insoluble in water but dissolve in acids to form salts containing the Pb2+ ion or in alkalies to form plumbites, which have the PbO22− ion. Litharge, which is produced by air oxidation of lead, is the most important commercial compound of lead; it is used in large amounts directly and as the starting material for the preparation of other lead compounds. Considerable quantities of PbO are consumed in manufacturing the plates of lead-acid storage batteries. High-quality glassware (lead crystal) contains as much as 30 percent litharge, which increases the refractive index of the glass and makes it brilliant, strong, and resonant. Litharge is also employed as a drier in varnishes and in making sodium plumbite, which is used for removing malodorous thiols (a family of organic compounds containing sulfur) from gasoline.
PbO2, found in nature as the brown-to-black mineral plattnerite, is commercially produced from trilead tetroxide by oxidation with chlorine. It decomposes upon heating and yields oxygen and lower oxides of lead. PbO2 is used as an oxidizing agent in the production of dyestuffs, chemicals, pyrotechnics, and matches and as a curing agent for polysulfide rubbers. Trilead tetroxide (known as red lead, or minium) is produced by further oxidation of PbO. It is the orange-red to brick-red pigment commonly used in corrosion-resistant paints for exposed iron and steel. It also reacts with ferric oxide to form a ferrite used in making permanent magnets.
Another economically significant compound of lead in the +2 oxidation state is lead acetate, Pb(C2H3O2)2, a water-soluble salt made by dissolving litharge in concentrated acetic acid. The common form, the trihydrate, Pb(C2H3O2)2•3H2O, called sugar of lead, is used as a mordant in dyeing and as a drier in certain paints. In addition, it is utilized in the production of other lead compounds and in gold cyanidation plants, where it primarily serves to precipitate soluble sulfides from solution as PbS.
Various other salts, most notably basic lead carbonate, basic lead sulfate, and basic lead silicate, were once widely employed as pigments for white exterior paints. Since the mid-20th century, however, the use of such so-called white lead pigments has decreased substantially because of a concern over their toxicity and attendant hazard to human health. The use of lead compound in insecticides has virtually been eliminated for the same reason.

It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1072 2021-07-06 01:01:44
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
1050) Mendelevium
Mendelevium (Md), synthetic chemical element of the actinoid series of the periodic table, atomic number 101. It was the first element to be synthesized and discovered a few atoms at a time. Not occurring in nature, mendelevium (as the isotope mendelevium-256) was discovered (1955) by American chemists Albert Ghiorso, Bernard G. Harvey, Gregory R. Choppin, Stanley G. Thompson, and Glenn T. Seaborg at the University of California, Berkeley, as a product resulting from the helium-ion (alpha-particle) bombardment of a minute quantity (about a billion atoms) of einsteinium-253 (atomic number 99). The element was named after Russian chemist Dmitry Mendeleyev.
In about a dozen repetitions of the experiment, the team of scientists produced 17 atoms of mendelevium, which were identified by the ion-exchange adsorption-elution method (mendelevium behaved like its rare-earth homologue thulium) and by the electron-capture decay of its daughter isotope fermium-256. Fifteen other isotopes of mendelevium, all radioactive, have been discovered. The stablest is mendelevium-258 (51.5-day half-life). Studied by means of radioactive tracer techniques, mendelevium exhibits a predominant +3 oxidation state, as would be expected by its position in the actinoid series; a slightly stable +2 oxidation state is also known.
Element Properties
atomic number : 101
stablest isotope : 258
oxidation states : +2, +3.
It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1073 2021-07-07 00:31:07
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
1051) Scandium
Scandium (Sc), chemical element, a rare-earth metal of Group 3 of the periodic table.
Scandium is a silvery white, moderately soft metal. It is fairly stable in air but will slowly change its colour from silvery white to a yellowish appearance because of formation of Sc2O3 oxide on the surface. The metal slowly dissolves in diluted acids—except hydrofluoric acid (HF), in which a protective trifluoride layer prevents further reaction. Scandium is paramagnetic from 0 K (−273 °C, or −460 °F) to its melting point (1,541 °C, or 2,806 °F). It becomes superconducting at −273.1 °C (−459.6 °F) at pressures exceeding 186 kilobars.
After Russian chemist Dmitry Ivanovich Mendeleyev in 1871 predicted this element’s existence, tentatively calling it ekaboron, Swedish chemist Lars Fredrik Nilson in 1879 discovered its oxide, scandia, in the rare-earth minerals gadolinite and euxenite, and Swedish chemist Per Teodor Cleve later in 1879 identified scandium as the hypothetical ekaboron. Scandium is found in small proportions, generally less than 0.2 percent, in many of the heavy lanthanide ores and in many tin, uranium, and tungsten ores. Thortveitite (a scandium silicate) is the only mineral containing large amounts of scandium, about 34 percent, but unfortunately this mineral is quite rare and is not an important source of scandium. The cosmic abundance of scandium is relatively high. Although it is only about the 50th most abundant element on Earth (its abundance is similar to that of beryllium), it is about the 23rd most abundant element in the Sun.
In nature, scandium exists in the form of one stable isotope, scandium-45. Among 25 (excluding nuclear isomers) radioactive isotopes with masses ranging from 36 to 61, the most stable is scandium-46 (half-life of 83.79 days), and the least stable is scandium-39 (half-life of less than 300 nanoseconds).
Scandium is separated from the other rare earths by precipitation of the insoluble potassium scandium sulfate or by extraction of scandium thiocyanate by diethyl ether. The metal itself was first prepared in 1938 by the electrolysis of potassium, lithium, and scandium chlorides in a eutectic mixture (i.e., a mixture having the lowest melting point possible with those components). Scandium is now produced mostly as a by-product of uranium extraction from the mineral davidite, which contains about 0.02 percent scandium oxide. Scandium exists in two allotropic (structural) forms. The α-phase is close-packed hexagonal with a = 3.3088 Å and c = 5.2680 Å at room temperature. The β-phase is body-centred cubic with an estimated a = 3.73 Å at 1,337 °C (2,439 °F).
Only a few uses of this unusual transition metal have been developed, mostly due to scandium’s limited availability and high cost. Its low density and high melting point suggest applications as an alloying agent for lightweight metals for military and high-performance applications. The major uses of scandium are as an alloy additive to aluminum-based alloys for sporting goods and in high-intensity metal halide lamps. When alloyed with aluminum and aluminum-based alloys, scandium limits high-temperature grain growth.
The chemistry of scandium bears a closer resemblance to that of the other rare-earth elements of oxidation state +3 than to that of aluminum or titanium. Some of its behaviour, however, is atypical of the rare earths because of its significantly smaller ionic radius (1.66 Å for coordination number 12) as compared with the rare-earth average (1.82 Å for coordination number 12). For this reason, the Sc3+ ion is a relatively strong acid and has a much greater tendency to form complex ions.
Element Properties
atomic number : 21
atomic weight : 44.95591
melting point : 1,541 °C (2,806 °F)
boiling point : 2,836 °C (5,137 °F)
specific gravity : 2.989 (24 °C, or 75 °F)
oxidation state : +3.

It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1074 2021-07-08 00:21:05
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
1052) Allotropy
Allotropy, the existence of a chemical element in two or more forms, which may differ in the arrangement of atoms in crystalline solids or in the occurrence of molecules that contain different numbers of atoms. The existence of different crystalline forms of an element is the same phenomenon that in the case of compounds is called polymorphism. Allotropes may be monotropic, in which case one of the forms is the most stable under all conditions, or enantiotropic, in which case different forms are stable under different conditions and undergo reversible transitions from one to another at characteristic temperatures and pressures.
Elements exhibiting allotropy include tin, carbon, sulfur, phosphorus, and oxygen. Tin and sulfur are enantiotropic: the former exists in a gray form, stable below 13.2° C, and a white form, stable at higher temperatures; sulfur forms rhombic crystals, stable below 95.5° C, and monoclinic crystals, stable between 95.5° C and the melting point (119° C). Carbon, phosphorus, and oxygen are monotropic; graphite is more stable than diamond, red phosphorus is more stable than white, and diatomic oxygen, having the formula O2, is more stable than triatomic oxygen (ozone, O3) under all ordinary conditions.
Allotropy (also known as allotropism) is when a chemical element can exist in two or more different forms in the same physical state or phase. It is the existence of a substance and especially an element in two or more different forms usually in the same phase. These different forms are called allotropes. The concept of allotropy was originally proposed in 1841 by the Swedish scientist Baron Jöns Jakob Berzelius (1779–1848).
Therefore, an allotrope is a different structure in which an element appears. They are different structural modifications of an element; the atoms of the element are bonded together in a different manner. This happens when the atoms of the element are bonded together in a different way. The ability for elements to exist in this way is called allotropism.
For example, the allotropes of carbon include:
a) diamond, where the carbon atoms are bonded together in a four-cornered lattice arrangement;
b) graphite, where the carbon atoms are bonded together in sheets of a six-sided lattice;
c) graphene, single sheets of graphite; and
d) fullerenes, where the carbon atoms are bonded together in spheres, cylinders or egg-shaped formations
Only some elements have allotropes.
Allotropes may display very different chemical and physical properties. The term allotropy is used for elements only, not for compounds. The more general term, used for any crystalline material, is polymorphism. For example, graphite and diamond are both allotropes of carbon that occur in the solid-state. Allotropy refers only to different forms of an element within the same state (i.e., different solid, liquid or gas forms); these different states are not, themselves, considered examples of allotropy. Phosphorus, sulfur, and tin also exhibit allotropy. It is a property of certain elements, as carbon, sulfur, and phosphorus, of existing in two or more distinct forms; allomorphism. Many metals have allotropic crystalline forms that are stable at different temperatures.
It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline
#1075 2021-07-09 00:31:17
- Jai Ganesh
- Administrator
- Registered: 2005-06-28
- Posts: 53,831
Re: Miscellany
1053) Krypton
Krypton (Kr), chemical element, rare gas of Group 18 (noble gases) of the periodic table, which forms relatively few chemical compounds. About three times heavier than air, krypton is colourless, odourless, tasteless, and monatomic. Although traces are present in meteorites and minerals, krypton is more plentiful in Earth’s atmosphere, which contains 1.14 parts per million by volume of krypton. The element was discovered in 1898 by the British chemists Sir William Ramsay and Morris W. Travers in the residue left after a sample of liquid air had boiled almost entirely away.
Element Properties
atomic number : 36
atomic weight : 83.80
melting point : -156.6 °C (−249.9 °F)
boiling point : -152.3 °C (−242.1 °F)
density (1 atm, 0 °C [32 °F]) : 3.733 g/litre (0.049 ounce/gallon)
oxidation numbers : 0, 2
Properties Of The Element
Because its boiling point (−152.3 °C, or −242.1 °F) is about 30–40 °C (50–70 °F) higher than those of the major constituents of air, krypton is readily separated from liquid air by fractional distillation; it accumulates along with xenon in the least volatile portion. These two gases are further purified by adsorption onto silica gel, redistillation, and passage over hot titanium metal, which removes all impurities except other noble gases.
Krypton is used in certain electric and fluorescent lamps and in a flashlamp employed in high-speed photography. Radioactive krypton-85 is useful for detecting leaks in sealed containers, with the escaping atoms detected by means of their radiation. Krypton is named from the Greek word kryptos, “hidden.”
When a current of electricity is passed through a glass tube containing krypton at low pressure, a bluish white light is emitted. The wavelength of an orange-red component of light emitted by stable krypton-86, because of its extreme sharpness, served from 1960 to 1983 as the international standard for the metre. (One metre equaled 1,650,763.73 times the wavelength of this line.)
Natural krypton is a mixture of six stable isotopes: krypton-84 (57.0 percent), krypton-86 (17.3 percent), krypton-82 (11.6 percent), krypton-83 (11.5 percent), krypton-80 (2.25 percent), and krypton-78 (0.35 percent). Krypton has isotopes of every mass number from 69 through 100; of these isotopes; twenty-five are radioactive and are produced by fission of uranium and by other nuclear reactions. The longest lived of these, krypton-81, has a half-life of 229,000 years. After it has been stored a few days, krypton obtained by nuclear fission contains only one radioactive isotope, krypton-85, which has a half-life of 10.8 years, because all the other radioactive isotopes have half-lives of 3 hours or less.
Compounds
Krypton is the lightest of the noble gases that form isolable chemical compounds in macroscopic amounts. For many years it was considered to be totally unreactive. In the early 1960s, however, krypton was found to react with the element fluorine when both are combined in an electrical-discharge tube at −183 °C (−297 °F); the compound formed is krypton difluoride, KrF2. Several other methods for the synthesis of KrF2 are now known, including irradiation of krypton and fluorine mixtures with ultraviolet radiation at −196 °C (−321 °F).
KrF2 is a colourless crystalline solid that is highly volatile and slowly decomposes at room temperature. No other molecular fluoride of krypton has been isolated, so all krypton compounds are derived from KrF2, where Kr is in the +2 oxidation state. Krypton difluoride is a powerful oxidative fluorinating agent. (Its oxidizing power means that it extracts electrons from other substances and confers on them a positive charge. Its fluorinating ability means that it transfers an F− ion to other substances. Hence, in a formal sense, oxidative fluorination is the net result of extraction of two electrons and addition of F−; this can be considered to be equivalent to the transfer of F+.) KrF2 is, for example, capable of oxidizing and fluorinating xenon to XeF6 and gold to AuF5.
The cationic species KrF+ and Kr2F3+ are formed in reactions of KrF2 with strong fluoride-ion acceptors such as the pentafluorides of Group 15, in which the fluoride ion F− is transferred to the pentafluoride to give complex salts that are analogous to those of XeF2; here no oxidation is involved. Among these complex salts are [KrF+][SbF6−] and [Kr2F3+][AsF6−]. The Kr2F3+ cation is V-shaped with a fluorine atom bonded to each of two krypton atoms and both krypton atoms bonded to a common fluorine in the middle, i.e., F(KrF)2+.
The KrF+ cation ranks among the most powerful chemical oxidizers presently known and is capable of oxidative fluorination of gaseous xenon to XeF5+ and chlorine, bromine, and iodine pentafluorides to the ClF6+, BrF6+, and IF6+ cations, respectively. The KrF+ cation behaves as only an oxidizing agent in converting gaseous oxygen to O2+.
The KrF+ cation has been shown to behave as a Lewis acid (electron-pair acceptor) toward a number of Lewis bases that are resistant to oxidation by the strongly oxidizing KrF+ cation at low temperatures. These Lewis acid-base adducts are exemplified by HCNKrF+ and F3CCNKrF+, which are formed as AsF6− salts. Such cations are the only known examples of krypton bonded to nitrogen. The compound Kr(OTeF5)2 is the only reported example of a compound in which krypton is bonded to oxygen. No compounds in which krypton is bonded to elements other than fluorine, oxygen, and nitrogen have been isolated.
Clathrate “compounds,” in which the element is trapped in cagelike structures of water or other molecules, are known. There is no diatomic molecule of krypton.

It appears to me that if one wants to make progress in mathematics, one should study the masters and not the pupils. - Niels Henrik Abel.
Nothing is better than reading and gaining more and more knowledge - Stephen William Hawking.
Offline